Size really does matter when sequencing the genomes of all life in Britain and Ireland
Published in Ecology & Evolution


Genome sizes vary wildly throughout nature. We explored some (perhaps unexpected) examples in our video, entered by the Darwin Tree of Life team in Nature’s Science in Shorts competition.
Genome size has tangible real-world consequences for different organisms in different environments. But it also has a big impact on how our scientists work.
The Darwin Tree of Life project aims to sequence and assemble the genomes of every plant, animal, fungus and single-celled protist in Britain and Ireland - perhaps some 70,000 species. The project involves ten partner organisations, with the bulk of the full genome sequencing happening at the Wellcome Sanger Institute in Cambridgeshire.
With so many varied species coming through our labs, knowing the size of their genomes is incredibly important for estimating the costs and computational power required, and for checking our work at the end.
Sizing up a genome
First, what do we mean by ‘genome size’? Put simply, it is the amount of DNA a species has in the nucleus of each cell. We measure this in base pairs - the ACGT code that forms the building blocks of DNA. The units used are usually either Mbp (mega base pairs, with 1 Mbp being a million base pairs) or Gbp (giga base pairs, with 1 Gbp being a billion base pairs). The human genome is roughly 3.2 Gbp.
Every plant species collected by the project first goes to the Royal Botanic Gardens, Kew. Here Dr. Ilia Leitch and her team measure the size of each plant’s genome using a process called flow cytometry.
 and the DToL team from Kew on a plant collecting trip on The Lizard with the Botanical Group Cornwall.")
“It’s deceptively simple really,” Ilia explains. “We take a fresh leaf and chop it up with a razor blade into a liquid. That breaks down the cells and releases the nuclei into the liquid, and we filter out everything else.
“Then we add a staining chemical that binds quantitatively to the DNA, meaning the more DNA there is in a nucleus the more stain will bind to it. Next we pass the nuclei through a machine called the flow cytometer, effectively a funnel with a very fine tube at the end. This channels all the nuclei into a single file. And, as they pass down the tube, the machine measures how much stain has bound to each nucleus - which tells you the genome size.”
The process needs to be done alongside the nuclei from another species for which the genome size is already known. This list of well-known species reads like a vegetarian cookbook as it includes pea (Pisum sativum), parsley (Petroselinum crispum), onion (Allium cepa), tomato (Solanum lycopersicum). By comparing the readings from the familiar species, the team can calculate the unknown species tested alongside it.
GoaT: Genomes on a Tree
The data from Ilia’s genome sizing at Kew gets uploaded to a powerful new genomics tool, developed by a team of bioinformaticians at Sanger. They named it ‘Genomes on a Tree’ - or ‘GoaT’ for short.
“GoaT is a really powerful aggregator, bringing together genome-relevant data from specialised and trusted sources everywhere, worldwide, into a single source,” explains Dr. Cibele Sotero-Caio, the curator of the data on GoaT.
Like a Google for genome data, users can put any species’ name into GoaT and receive its genomic vitals. This includes information on whether that genome has been assembled before, the quality of that assembly, the number of chromosomes, and the genome size.
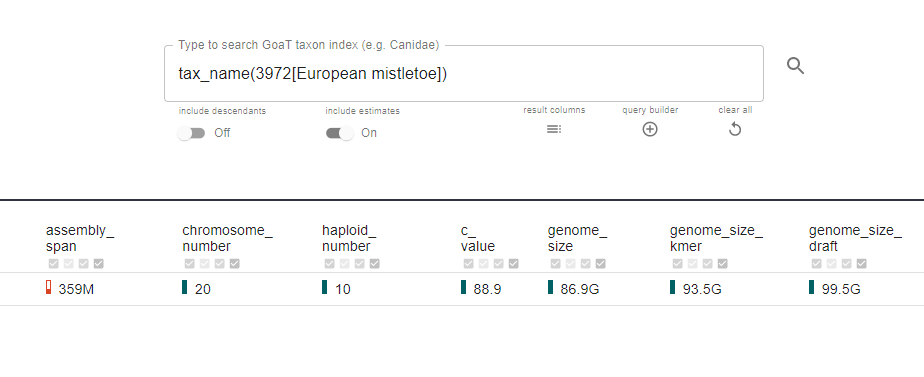
For species with an unknown genome size, GoaT uses estimates based on the genome sizes of the species’ closest relatives, and a clear colour-coding system to show how reliable the data is.
Phase one of the Darwin Tree of Life project is prioritising the sequencing of representatives of each family of organisms in Britain and Ireland. There are approximately 9,000 families of species across the globe, 4,000 of which live here.
“We’ll have at least one genome size estimate per family soon, which will help correct the rest of the data,” explains Cibele. “You can actually think about GoaT in the same way we think about science. Science corrects itself as we learn more. Over time we know GoaT estimates will also get better.”
Incidentally, a goat (Capra hircus) has a genome roughly the same size as ours.
Sequencing mistletoe in time for Christmas
Once the DNA has been extracted from a sample at Sanger it is sent to the long-read sequencing team, who specialise in producing longer DNA sequence data. Essentially, physical DNA is transformed into large computer files full of ACGT code. This is done by powerful machines called Sequel IIe systems, made by California-based company Pacific Biosciences (PacBio).
“We’re doing the physical work of actually running the sequencing,” explains James Watts, who leads one of the long-read teams. “We take the DNA and make it machine readable, so it can be processed.”
At the heart of the process is something called a SMRT cell (pronounced ‘smart’). Every machine can hold four cells, each of which have 8 million pores. “Each cell is really quite tiny. It fits in the palm of your hand. Essentially you have 8 million chances to read the DNA you load onto each cell,” explains James, and this provides enough ‘coverage’ to make an attempt at a high-quality genome assembly.

Here too, knowing genome size is crucial. “The data from GoaT feeds heavily into our strategy and approach. Anything with a genome size less than 1 Gbp, we know we’ll be able to adequately cover that with one smart cell in about one day.”
Towards the end of 2021, the team received DNA from the mistletoe. This hemiparasitic yuletide favourite has the largest genome of any species living wild in Britain and Ireland - more than 90 Gbp. James and the team ran all 12 machines for a week to get the coverage of DNA data they needed.
“It wasn’t on purpose, but we found ourselves racing to sequence the mistletoe before Christmas. We knew that meant a lot of smart cells. So there was a bit of excitement there, and some relief once we’d completed it.”
Putting the genomic jigsaw back together
The act of extracting and sequencing DNA breaks it up into fragments. The code must be put back in the correct order, reflecting its position along the different chromosomes. This is where the Tree of Life Assembly team comes in.
“Knowing the genome size ahead of time is helpful for knowing the kind of compute resource you’re going to use. For mistletoe, for instance, we did a lot of special things because we knew the genome would be so large,” explains Dr. Shane McCarthy who heads the team.

For mistletoe, three terabytes of storage was needed just to get the raw data on disk. Only two machines at Sanger had the memory to actually do the assembly. Then, to map the genome, the team has lots of small jobs running in parallel on different computers. “For a smaller genome you might have 10 jobs running. For the mistletoe it was hundreds,” says Shane.
Again, Shane’s team refers back to the GoaT database. But they now also have the sequence data, provided by the long-read team and their PacBio machines. A software called GenomeScope provides estimates based on the sequence data. This is then cross-referenced with GoaT to check if the figures add up or whether more data needs to be generated before an attempt on the assembly is made.
Genome size data is also used to check how complete the final assembly is. “You want your assembly to reflect the true genome size,” says Shane.
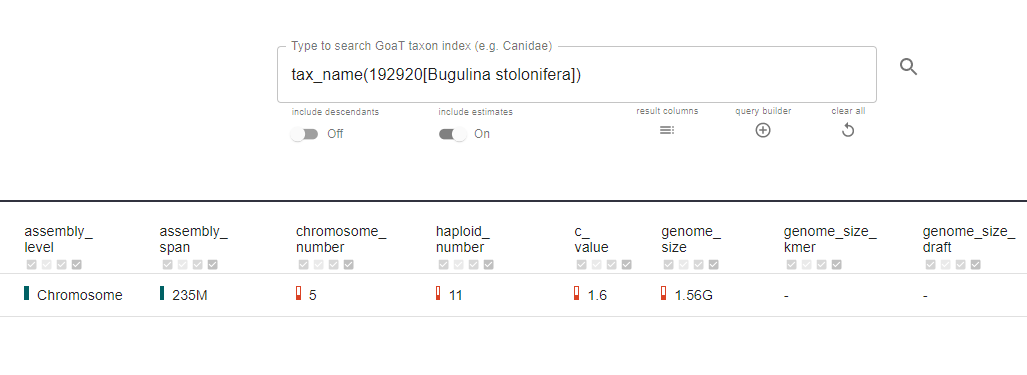
However, the figures in GoaT and the final assembly don’t always match, especially for more obscure parts of the tree of life. Shane points to a bryozoan, Bugulina stolonifera, which GoaT suggested had a genome size of 1.5 Gbp. In fact, once assembled, the size was six times smaller.
But remember, many of the genome sizes in GoaT are estimates based on the closest relatives with known genome assembly data. What’s more, biology occasionally throws up surprises - even between closely related species.
In the three decades Ilia Leitch has been studying genome size, her biggest surprise came from an Australian fern, Tmesipteris obliqua. It had a genome size of almost 150 Gbp, double any other fern.
“It’s really exciting to be involved with the Darwin Tree of Life project,” says Ilia. “These high-quality reference genomes we’re producing will provide us with exquisite detail, not only into how genes are organised - which is what a lot of people are interested in - but also how the non-coding DNA is organised and what this means for the species.”




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in