Super Trouper Resolution: Fusing Histology with Spatial Expression Data for Super-Resolved Transcriptomics
Published in Bioengineering & Biotechnology

Histology, the study of microscopic tissue structures, has been a central component in biological research ever since the 17th century [1]. As anyone who has ever had the chance to study a biological sample using a microscope can attest to, the amount of information present in just a few millimeters of tissue can be staggering. By studying how tissues organize, it is possible to explain not only how they function, but also to identify common disease states. Indeed, the same methods pioneered hundreds of years ago are still routinely used today to diagnose and treat a wide range of ailments.
In recent years, the advent of spatial transcriptomics has allowed researchers to quantify localized gene expression in order to improve our understanding of how cells communicate and mutually affect each other in a spatial context. Historically, spatial analyses of transcript expression have relied on hybridizing labeled probes to preselected transcript targets [2]. However, since only a limited number of transcripts can be targeted at once, these methods necessitate prior knowledge of the process being studied and its transcriptomic drivers. Although considerable progress has been made in increasing the number of transcript targets, hybridization-based spatial transcriptomics remains prohibitively costly and difficult to use in exploratory research when targets are unknown.
Today, new powerful methods for spatial transcriptomics have been developed that can quantify the expression of all protein-coding genes simultaneously [3], highlighted by the recent announcement of spatially resolved transcriptomics as the Method of the Year 2020 [4]. These methods are based on capturing transcripts in barcoded tissue regions. While enabling untargeted gene expression quantification, they typically have lower resolution and insufficient sensitivity to robustly characterize minute anatomical features. Thus, researchers are caught between a rock and a hard place in that they must decide between either quantifying the expression of only a limited set of genes using hybridization-based spatial transcriptomics or quantifying the expression of all protein-coding genes but at a lower sensitivity and resolution using capture-based spatial transcriptomics [5].
When we initially set out to study how histology could be utilized in spatial transcriptomics experiments, we used deep learning to predict the gene expression in a capture area from a high-resolution hematoxylin and eosin stain of the same region. Our results, published last year in Nature Biomedical Engineering [6], showed that it was possible to predict the expression of breast cancer markers with a reasonable degree of confidence. Moreover, we could attribute predictions to particular histological features within the capture area. Encouraged by this result, we hypothesized that it may be possible to leverage histology to impute transcript abundances and thus endow capture-based spatial transcriptomics with much higher resolution, ameliorating its disadvantages compared to hybridization-based methods.
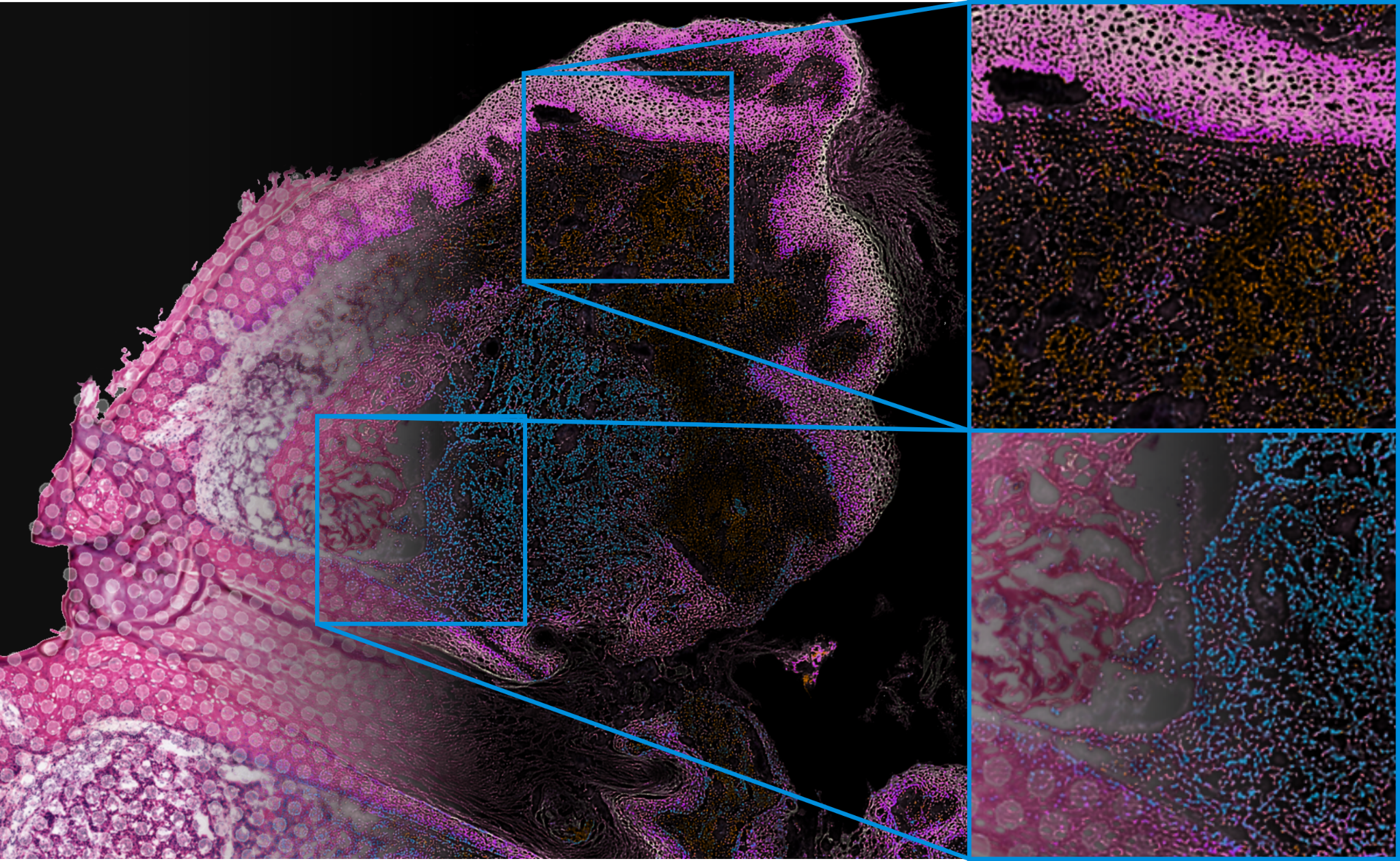
In our paper published today in Nature Biotechnology, we present a method for integrating gene expression data from capture-based spatial transcriptomics with histology images. The proposed model enforces a tight coupling between gene expression and histology, making it possible to infer full-transcriptome, pixel-level transcript abundances. We show that the method can predict gene expression not only within and between the original capture areas but also in neighboring sections for which only histological images are available. The method can be used to characterize the transcriptome of micrometer-scale anatomical features and to reduce sequencing needs by imputing expression using reference experiments.
From a broader perspective, our work suggests that histology is a vastly underutilized resource in spatial transcriptomics. Histology images have high resolution and negligible measurement noise. Using multi-modal learning and domain adaptation, it is possible to transfer those qualities to spatial transcriptomics data and learn integrative tissue models. The method that we present in our paper is a first step in this direction, but much more is possible. In the future, we expect to see even larger-scale tissue models, leveraging the distinct qualities of data from spatial transcriptomics, histology, and perhaps other modalities [7]. Such models could, for example, characterize the genetic drivers of morphology or be used as a basis for explainable AI in digital pathology.
References
[1] Bracegirdle, B. The History of Histology: a Brief Survey of Sources. History of Science 15, 77–101 (1977). URL https://doi.org/10.1177/007327537701500201.
[2] Femino, A. M. Visualization of Single RNA Transcripts in Situ. Science 280, 585–590 (1998). URL https://doi.org/10.1126/science.280.5363.585.
[3] Ståhl, P. L. et al. Visualization and Analysis of Gene Expression in Tissue Sections By Spatial Transcriptomics. Science 353, 78–82 (2016). URL https://doi.org/10.1126/science.aaf2403.
[4] Larsson, L., Frisén, J. & Lundeberg, J. Spatially Resolved Transcriptomics Adds a New Dimension To Genomics. Nature Methods 18, 15–18 (2021). URL https://doi.org/10.1038/s41592-020-01038-7.
[5] Asp, M., Bergenstråhle, J. & Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. BioEssays 42, 1900221 (2020). URL https://doi.org/10.1002/bies.201900221.
[6] He, B. et al. Integrating Spatial Gene Expression and Breast Tumour Morphology Via Deep Learning. Nature Biomedical Engineering 4, 827–834 (2020). URL https://doi.org/10.1038/s41551-020-0578-x.
[7] Erickson, A. et al. The spatial landscape of clonal somatic mutations in benign and malignant tissue (2021). URL https://doi.org/10.1101/2021.07.12.452018.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in