Systematic and Reversible Control of Linear Conjugation along Polymer Backbones
Conjugated polymers contain unsaturated units that extend π-electron delocalization along the backbone, endowing them with tunable semiconducting properties absent in saturated, insulating polymers. Decades of molecular engineering have refined their electronic and optical behavior for plastic electronics, yet reversibly toggling their backbones between wide-bandgap insulators and conventional semiconductors remains a key unmet goal.
Herein, we present a novel, general strategy to create polymer architectures capable of dynamically switching between insulating and semiconducting states through reversible molecular reconfiguration with bistability. This strategy involves copolymerizing readily accessible lactone-functionalized xanthene moieties with traditional π-conjugated units (Fig. 1a). Upon acid or electrical stimuli, linear conjugation can be induced systematically along polymer backbones, either stepwise or synergistically, allowing tunable π-orbital overlap. Given the extensive applications of conjugated polymers, this reversible conjugation approach provides promising routes toward next-generation smart materials.

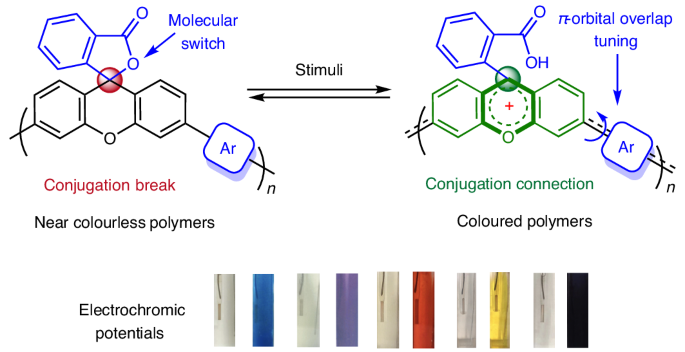
Figure 1. Proposed macromolecular structure enabling reversible control of conjugation in response to external stimuli and working mechanism for xanthene dyes.
Our design was guided by two key challenges: (i) identifying robust frameworks capable of reversible conjugation switching upon stimuli and (ii) developing effective control strategies. Inspired by pH-responsive xanthene dyes like fluorescein and rhodamine (Fig. 1b), we integrated lactone-functionalized xanthene units into polymer backbones. Upon protonation, the spirocyclic sp³ center converts to a planar sp² carbocation, opening a π-conjugation pathway between the 3′ and 6′ positions. Deprotonation restores the lactone, disrupting conjugation. This reversible switching enables dynamic modulation of conjugation via C1-centered carbocation intermediates, propagating local changes across the polymer chain.
Systematic control over linear conjugation was realized through strategic comonomer unit design. Polymer 3a, synthesized from 3′,6′-dibromo-xanthene (1a) and thieno[3,4-c]pyrrole-4,6-dione (2a), showed stepwise color changes upon acid/base stimuli, reflecting progressive modulation of conjugation (Fig. 2a). To tune π-orbital overlap along the backbone, we evaluated several monomers, 3,4-propylenedioxythiophene (2b), 2,1,3-benzothiadiazole (2c), its derivatives (2c′), and 1,2,4,5-tetrafluorobenzene (2d), as copolymer partners with xanthene derivatives (1a or 1b). Resulting polymers (3b–3d) exhibited blue, magenta, red, and yellow hues upon TFA treatment, confirming band-gap tunability (Fig. 2b). Additionally, random copolymers 3e–3g were synthesized via Pd-catalyzed direct arylation polymerization using mixed comonomers, producing switchable orange, green, and black colors under acidic conditions (Fig. 2c). Unlike the stepwise switching of 3a, polymers 3b–3g displayed synergistic, full-spectrum color changes upon stimuli.
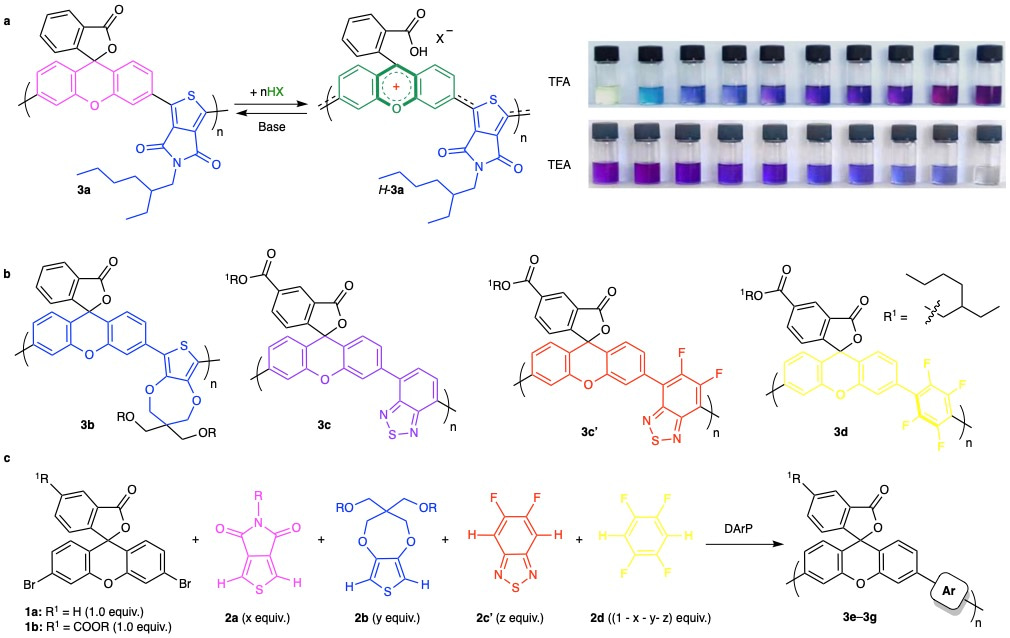
Figure 2. Synthesis and reversible control of the conjugation-tunable polymer.
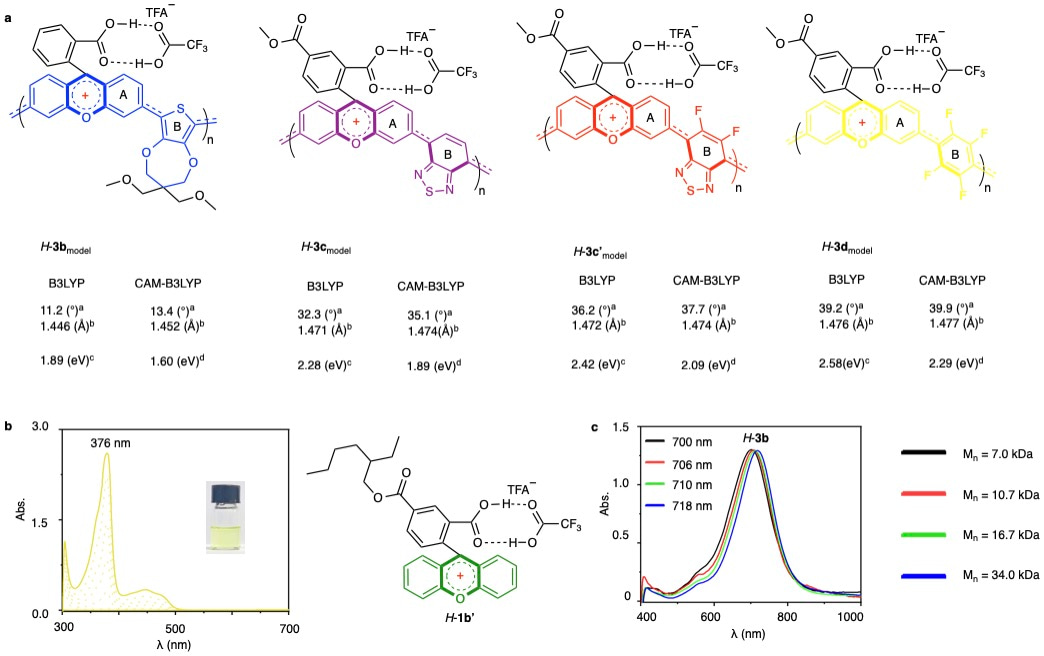
Beyond their reversible colorless-to-colored switching behavior that renders them promising for electrochromic (EC) devices, these materials were examined further for structure-property relationship by density-functional-theory (DFT) calculations and complementary mechanistic studies (Fig. 3). Optimized structures for truncated models of H-3b, H-3c, H-3c', and H-3d at the B3LYP/6-31G(d,p) level revealed carbon–carbon bond lengths between aromatic units ranging from 1.446 to 1.476 Å. These values fall between those of typical single and double bonds, suggesting the presence of partial π-conjugation (Fig. 3a). These bond lengths correlated with increasing dihedral angles (11.2° to 39.2°) between xanthene and adjacent aromatic units, reflecting decreasing planarity. Parallel CAM-B3LYP calculations followed these trends. Upon acid stimulation, the xanthene-centered positive charge delocalized onto neighboring aromatic units, as further supported by ESP maps and a linear relationship between electrostatic potential and dihedral angle. This demonstrates that tuning the electronic properties of aromatic comonomers enables control over π-orbital overlap and band-gap modulation. DFT-calculated band gaps closely matched those estimated from UV/Vis spectra, while TDDFT simulations reproduced λmax values consistent with experimental data for H-3b–H-3d. To further validate conjugation extent, the small molecule analog 1b’ was synthesized and treated with TFA. It displayed an absorption peak at 376 nm (Fig. 3b), significantly shorter than polymer H-3d (460 nm), confirming extended conjugation in polymeric structures, even in highly electron-deficient units. Molecular weight effects on optoelectronic properties of polymer 3b were also examined. While solution-phase UV/Vis absorption remained constant across varying molecular weights (7.0–34.0 kDa), thin-film devices exhibited a notable red shift (700 nm to 718 nm) upon electrical stimulation (Fig. 3c). This confirms extended linear conjugation across polymer chains of up to approximately 40 repeat units in the solid state.
Through copolymerizing readily accessible lactone-functionalized xanthene moieties with traditional π-conjugated units, we successfully developed polymeric architectures capable of dynamically switching between insulating and semiconducting states through reversible molecular reconfiguration. We hope that this work will stimulate further research interest in developing stimuli-responsive smart materials for plastic electronics.
For more details of this work, please see the original paper entitled “Reversible Formation and Control of Linear Conjugation in Polymers” in Nature Chemistry https://www.nature.com/articles/s41557-025-01851-7 . For more information on please refer to our group website at https://www.x-mol.com/groups/kai_lang?lang=en.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in