Taking peptides as a pill
Published in Bioengineering & Biotechnology

As an active member of the field of peptide engineering and drug development for over a decade now, the question that I have been asked the most is: "Can (bi)cyclic peptides be applied orally?" To me, this question has always been the most interesting, and yet also the most challenging. Peptides are notoriously unstable to proteases: how could peptides be evolved that survive the extremely high protease pressure in the gastrointestinal tract?
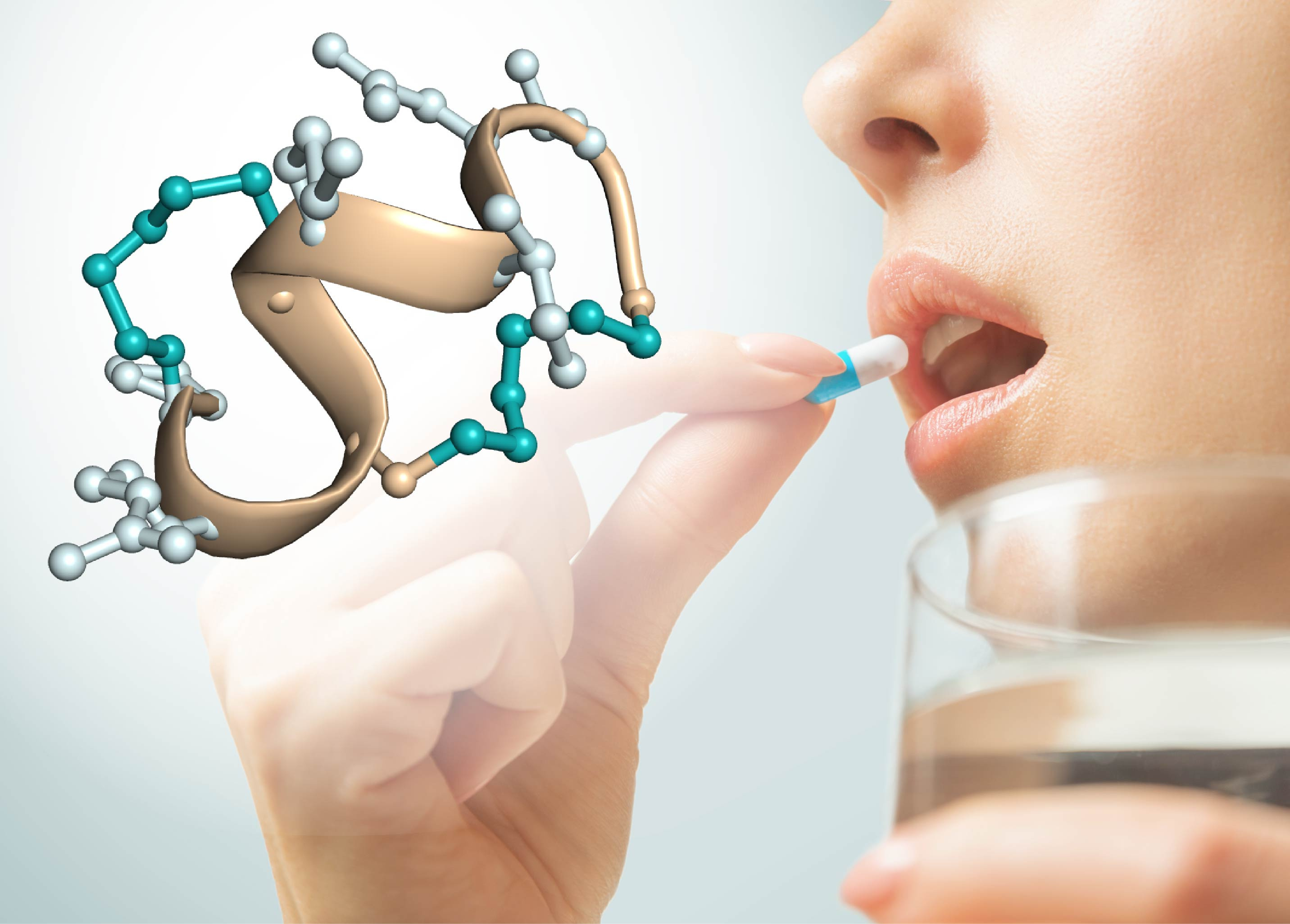
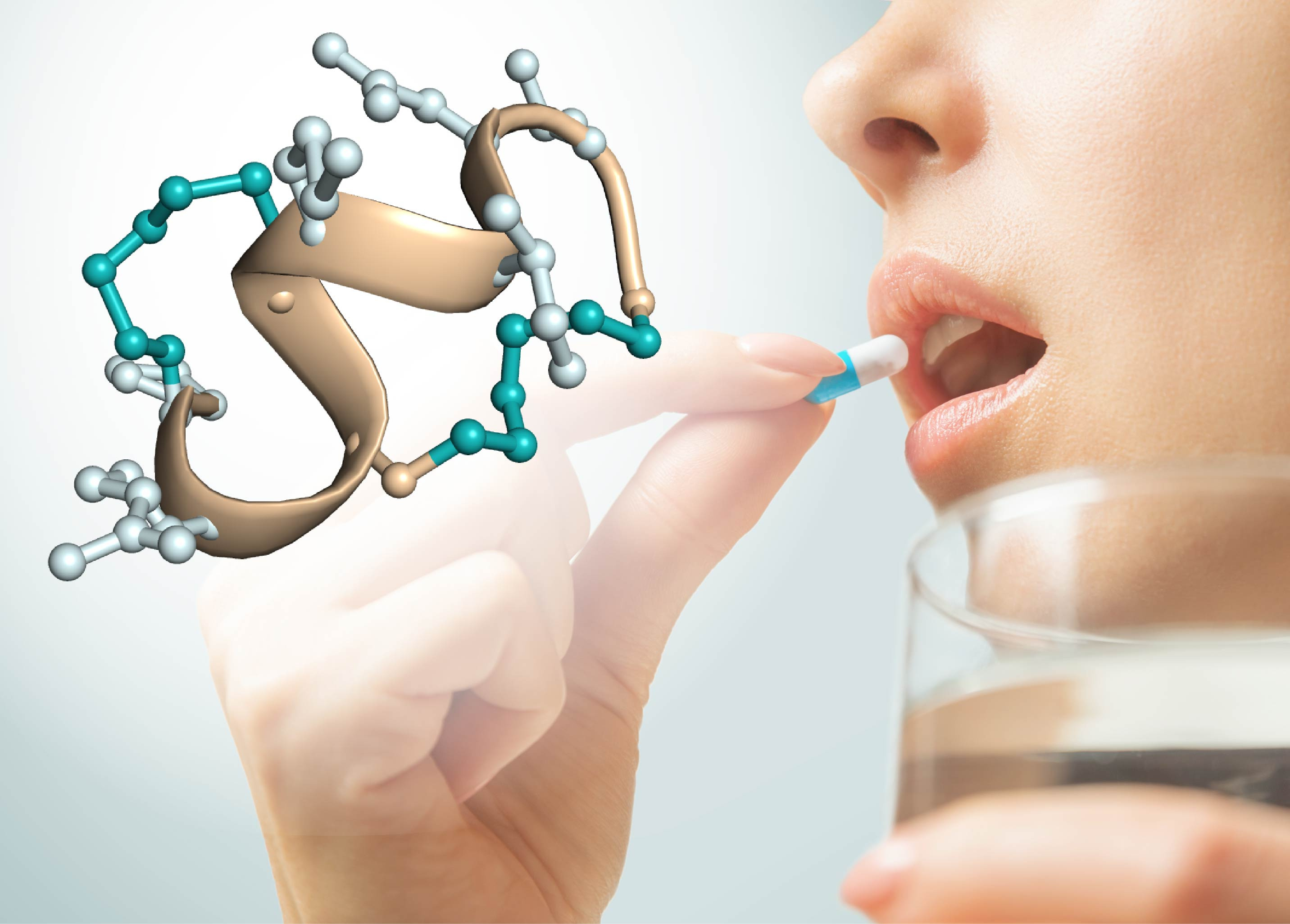
Figure 1: Oral administration of peptide drugs. Structure
of a protease-stable double-bridged peptide evolved by phage display.
At the end of the 1990's, an elegant method for evolving protease-stable proteins by phage display was published independently by two groups.1,2 In this strategy, protein libraries were displayed on phage, exposed to proteases, and intact (and thus stable) proteins were isolated by affinity selection. I found this approach highly attractive, and it appeared to be compatible with bicyclic peptide phage display, a method that Sir Greg Winter and I had developed to generate bicyclic peptide ligands.3
In my lab’s first attempt at this approach, post-doc collaborator Vanessa Baeriswyl (now at Janssen) incubated our phage library, which displays peptides constrained into bicycles on the phage surface, with proteases from cow intestine prior to the affinity selection. She was able to enrich for peptides that were substantially more stable than usual.4 However, the peptides were not good enough for oral application, as they could only survive cow proteases that were diluted more than 100 fold.
At this point, we were unsure if there were simply no sufficiently stable bicyclic peptides in the library or if the problem was the limited stability of the phage particle itself. The phage for this library were mutated to be disulfide free to facilitate the peptide bi-cyclization reaction via cysteines, however, this made the phage less stable towards proteases.
In the following years, our attempts to make protease-resistant peptides were put on hold. However, one of Vanessa’s experiments was stuck in my head — she showed that the non-mutated, wildtype phage was completely resistant to undiluted cow intestinal proteases.4 In the following years, Sangram Kale, a former post-doc who is now at Pepscan (the company where the chemistry for the bicyclic peptides was born) and Camille Villequey, a former PhD student (now at Novo Nordisk), found that peptides based on a new format termed "double-bridged peptides" can have an intrinsically high proteolytic stability.5 This finding re-activated our hope that super-stable peptides could be obtained.
With Xudong Kong, a current post-doc in the lab, we decided to combine the protease-stable, wildtype phage and the new double-bridged peptide format. A challenge with the wildtype phage was that it contains three disulfide bridges in an essential coat protein (protein 3), and care had to be taken that they were not damaged in the chemical reactions to convert the displayed linear peptides to double-bridged ones. Xudong tackled this problem along with several other formidable technical challenges — as described in the paper — and managed to generate a first completely artificial peptide ligand (Figure 1) that survived the harsh gastrointestinal protease pressure when fed to mice.
Key for this project’s success was Xudong’s talent for innovating efficient strategies for the parallel handling of many of the processes, such as cyclizing peptides with a large panel of chemical linkers, phage panning under numerous different conditions, and multiplexing solutions for DNA sequencing. An example of this is illustrated by the home-made magnetic array that Xudong used to perform dozens of affinity selections in parallel (see Figure 2).
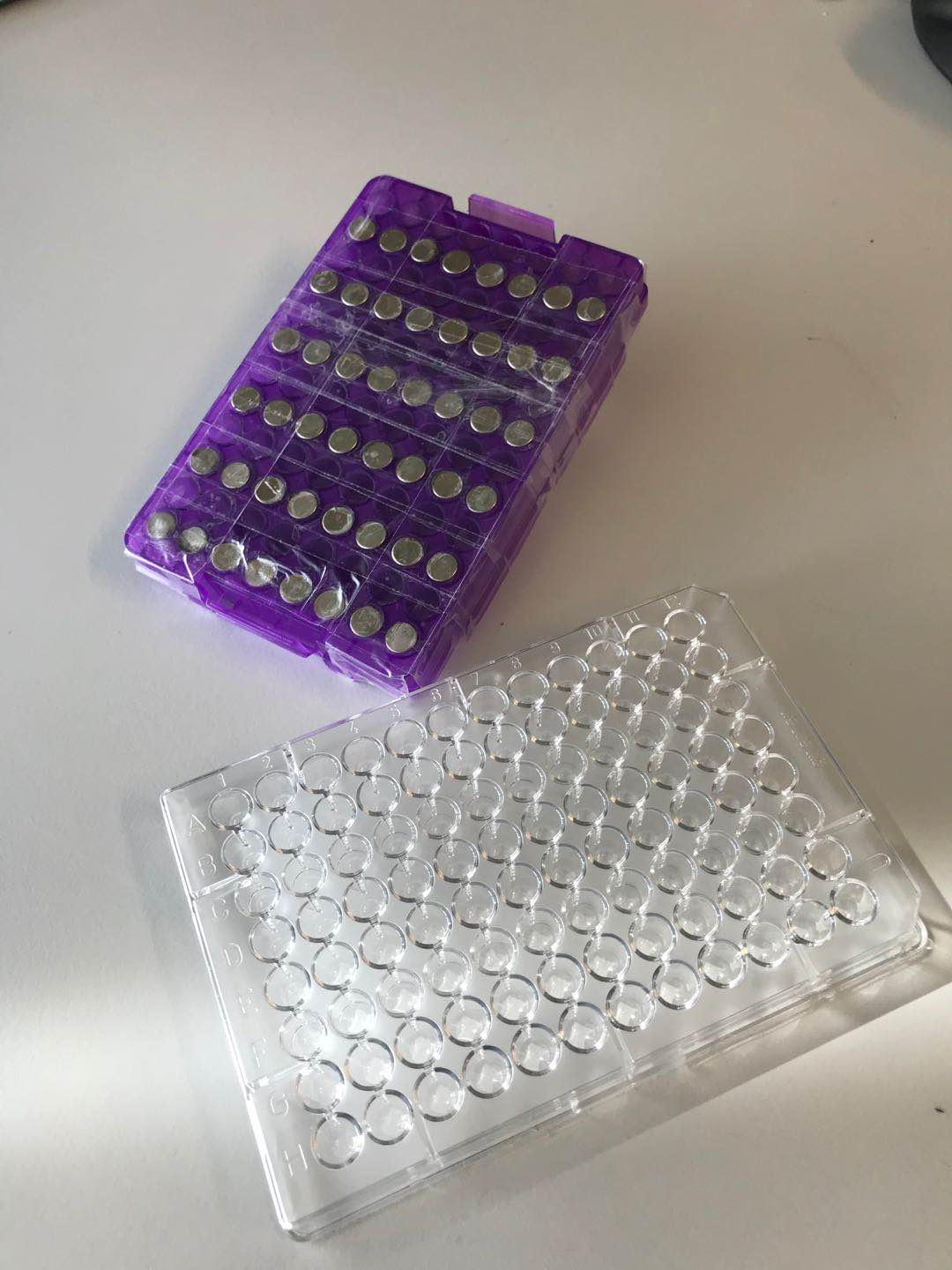
Figure 2: High-throughput is key. Home-made magnet array
used for phage display selections.
In this work, much of the peptide remained intact in the intestine of mice, which was our goal. However, only a small fraction made it to the blood, which highlights the next challenge for orally available peptide drugs — crossing the intestinal epithelium. While we are currently addressing this challenge as well, in parallel, we are using our current technology for developing protease-stable peptides to inhibit local targets in the intestine for which the peptides do not need to enter the blood stream. A visiting scientist from the Japanese company Eisai, Jun Moriya, applied this method and successfully generated protease-stable peptides against the IL-23 receptor, as described in the last part of the paper, and we hope that they can be translated into an oral peptide therapeutic.
References:
1. Kristensen, P. & Winter, G. Proteolytic selection for protein folding using filamentous bacteriophages. Fold. Des. 3, 321–328 (1998).
2. Sieber, V., Plückthun, A. & Schmid, F. X. Selecting proteins with improved stability by a phage-based method. Nat. Biotechnol. 16, 955-960 (1998).
3. Heinis, C., Rutherford, T., Freund, S. & Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 5, 502–507 (2009).
4. Baeriswyl, V. & Heinis, C. Phage selection of cyclic peptide antagonists with increased stability toward intestinal proteases. Protein Eng. Des. Sel. 26, 81-89 (2013).
5. Kale, S. S. et al. Cyclization of peptides with two chemical bridges affords large scaffold diversities. Nat. Chem. 10, 715–723 (2018).
Follow the Topic
-
Nature Biomedical Engineering
This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.

Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Medical Ultrasound: Emerging Techniques and Applications
Publishing Model: Hybrid
Deadline: Jan 29, 2027

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in