Targeted metabolomic analysis in Parkinson’s disease brain frontal cortex and putamen with relation to cognitive impairment
Published in Neuroscience

Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder. The disease is characterized by loss of motor control due to death of brain neurons connecting substantia nigra and putamen that produce neurotransmitter dopamine. Non-motor symptoms also frequently appear, including dementia, for which PD patients are at 6× higher risk than comparable non-PD population. Currently, there is no cure for the disease. Current therapies, such as exogenously supplied levodopa (L-DOPA; l-3,4-dihydroxyphenylalanine), a precursor of dopamine, can reduce motor symptoms but only temporarily and it gradually loses effectivity during the course of the treatment. The progressively disabling nature of PD results in a diminishing quality of life of the patients and a high burden for society.
While accumulation of α-synuclein protein aggregates, known as Lewy bodies, in brain is a well-known hallmark of PD, the underlying mechanisms triggering the pathology or development of dementia remain unknown, except for certain genetic mutations that account for around 5% of sporadic cases. Various systems biology approaches (“omics”) have been applied in the past to investigate the disease etiology. Metabolomics is a scientific field that studies the functional part of an organism, which is closest to the resulting phenotype, by measuring metabolites – small molecules and lipids that arise during metabolic processes. Recent rapid technological developments in this area have allowed large scale metabolomic studies, that can potentially quantitate hundreds or thousands of metabolites simultaneously.
In our current case-control study with 101 subjects, we performed a large explorative targeted metabolomic analysis directly in human post-mortem brain tissue – frontal cortex and putamen – encompassing the main metabolic pathways as well as certain lipid classes, to better understand brain metabolic changes associated with PD at various stages of cognitive impairment and the immediate effect of levodopa medication. This is the first study in human PD brain of its scope and its quality is underlined by very short post-mortem tissue collection intervals – averaging only 3 hours – with homogeneous distribution between the subject groups, which is highly important due to continuing post-mortem decay.
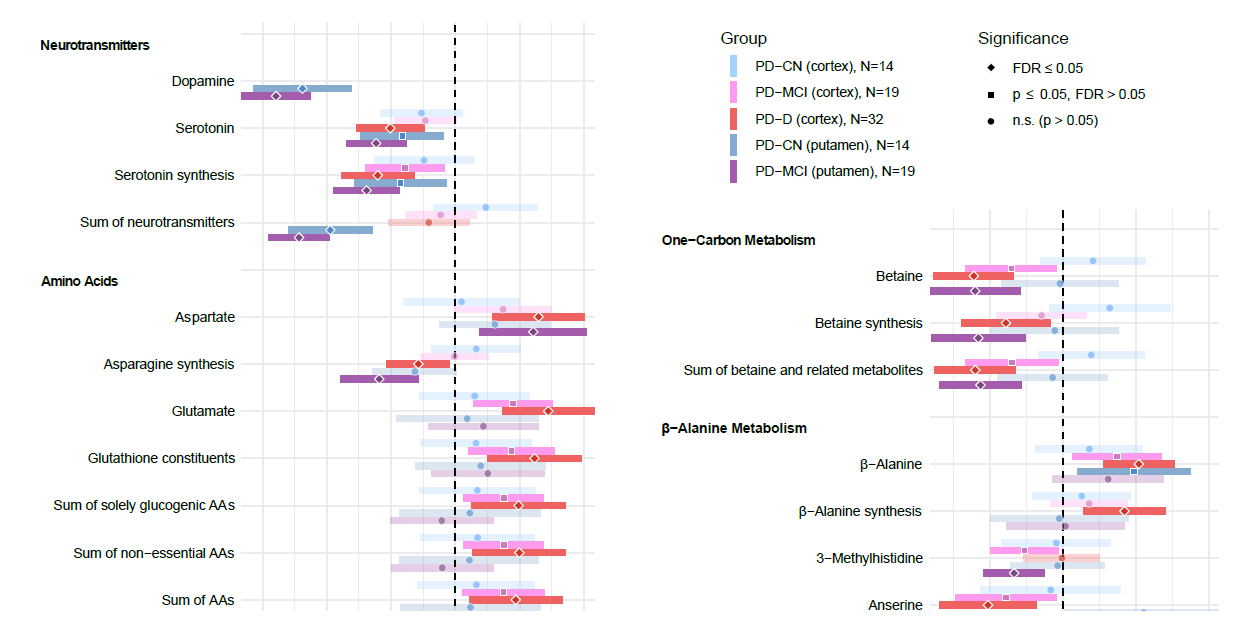
Our results reveal altered concentrations of metabolites across multiple pathways in association with:
- PD diagnosis – neurotransmitters, secondary bile acids
- Specific cognitive subgroups in PD – glutamate metabolism, polyamines, betaine metabolism
- Acute levodopa presence – one-carbon metabolism, lysophosphatidylcholines, cholesteryl esters, acylcarnitines, tricarboxylic acid cycle, β-alanine metabolism; some levodopa-related changes further exhibit an apparent temporal delay in PD with dementia – polyamines, fatty acids, phosphatidylcholines, glycosylceramides
- Clinical and histological scores of disease progression – neurotransmitters, primary bile acids, amino acids, betaine metabolism, carboxylic acids, acylcarnitines
While some changes have already been well documented, e.g. disturbance in the production of the neurotransmitter serotonin, other findings are novel or put into new perspective. The most important ones are summarized as follows:
PD subjects had an elevated indicator of glycine conjugation of deoxycholic acid, which is a ratio of glycodeoxycholic acid over deoxycholic acid. These secondary bile acids are derived from primary bile acids by microbial activity. Their detection in brain tissue itself represents another evidence for the existence of a gut-brain interaction. The perturbation of the secondary bile acids in PD regardless of disease progression suggests that microbiome might play a role in the etiology of the disease.
Cognitive impairment in PD (including both mild cognitive impairment and dementia) was associated with increased amino acids glutamate and aspartate. Glutamate accumulation is neurotoxic and its involvement in PD has long been suspected. Glutamate in brain is constantly metabolized and recycled, is inconvertible with aspartate, and can be unidirectionally exported through the blood-brain barrier into blood (both amino acids are unable to cross the barrier in the opposite direction from blood). We propose that the glutamate export is partially inhibited by observed increased concentration of α-aminoadipate (Fig. x1), as this compound negatively regulates cystine/glutamate antiporter, which exports glutamate. The elevation of α-aminoadipate might be due to dysfunction of 2-oxoadipate dehydrogenase that works downstream of α-aminoadipate. This dehydrogenase shares several subunits with 2-oxoglutarate dehydrogenase, which is a part of tricarboxylic acid cycle that processes 2-oxoglutarate – another metabolite interconvertible with glutamate. We hypothesize that a single cause can disrupt function of both dehydrogenases, effectively reducing glutamate clearance and metabolism, leading to its accumulation and contributing to cognitive impairment. Such a failure might be caused by an inadequate amount of one of several B vitamins that the dehydrogenases depend on.

Fig. x1 Glutamate metabolism and related pathways altered in PD with cognitive impairment. Selected metabolic reactions pertaining to glutamate metabolism and its connection with α-aminoadipate and observed changes. Arrows: red – upregulated; blue – downregulated; black solid – metabolic reaction; black sparsely dashed – export from brain; black densely dashed – inhibition. Font: gold – measured metabolite; gray – metabolite not included in the assay; purple – enzymes and cofactors; bright blue – transporters. Abbreviations: 2-AA α-Aminoadipate, 2-OA 2-Oxoadipate, 2-OG 2-Oxoglutarate, Asp Aspartate, Bn Active form of vitamin Bn, CoA coenzyme A, DH Dehydrogenase complex, Glu Glutamate, Lys Lysine, NAA N-Acetylaspartate, OA Oxaloacetate, Orn Ornithine, Suc Succinic acid, TCA Tricarboxylic acid, xCT Cystine/glutamate antiporter.
When we focused on the distinction between PD subjects with dementia and without dementia, we found no better discriminator than levodopa-related homocysteine accumulation that we previously reported (PubMed ID 35276958). Homocysteine is a toxic byproduct of the methylation cycle that has been previously associated with cognitive impairment and atherosclerosis. It is known that a part of the PD medication levodopa can be metabolized by methylation, producing homocysteine. However, nobody has shown how much this adverse processing occurs in human brain. We previously found that when levodopa was present in the brain tissue, only PD subjects with dementia had high levels of homocysteine (Fig. x2). In this study, we did not see any better discriminator of PD dementia in other metabolic pathways, strengthening the role of levodopa-related homocysteine accumulation as a probable cause of dementia in PD. Enzymes in homocysteine metabolism and clearance rely on several B vitamins and betaine (trimethylglycine).

Fig. x2 Levodopa-associated homocysteine elevation in PD with dementia. This box plot shows homocysteine concentrations across subject groups (CN – cognitively normal; MCI – with mild cognitive impairment; D – with dementia) in dependence on the acute levodopa medication presence (L+; red points) or physiological range (L-; black points). Note the large levodopa-associated homocysteine increase specific for PD-D group (p = 5e-12, FDR = 2e-10). The ROC curve and AUC refers to the classification of dementia status of PD subjects in L+ state.
We discovered many other associations with acute levodopa presence, confirming that it may play a substantial role in modulation of the metabolism beyond dopamine restoration. Surprising is the finding of apparent phase delay in the levodopa cyclical effect in cortex of PD subjects with dementia, for whom some changes are visible during normal DOPA levels instead of during acute levodopa medication presence (Fig. x3). It is uncertain whether this is due to a downstream effect of the differential homocysteine processing or another affected regulatory mechanism. This phenomenon deserves further validation and study.

Fig. x3 Example of phase delay in levodopa effect in PD with dementia. In this box plot (center line – median; box limits – upper and lower quartiles; whiskers – 1.5× interquartile range), depicted across subject groups (CN – cognitively normal; MCI – with mild cognitive impairment; D – with dementia of non-Alzheimer’s type) in dependence on the acute levodopa medication presence (L+; red points) or physiological range (L-; black points), we show an example of a metabolite (phosphatidylcholine PC aa C38:4; normalized values) with a seemingly opposite levodopa effect in PD-D subjects (decreasing red arrow). However, the values do not go in the opposite direction (below the baseline), but rather the same change happens in the opposite levodopa state (increase in L- instead of a change in L+), suggesting a delay in the levodopa-induced effect in PD-D cortex, potentially starting to appear in PD-MCI cortex. Similar patterns were observed across multiple metabolic classes. Statistical significance of regression coefficients for acute levodopa presence (from the main regression analysis, which includes covariates) is provided.
In conclusion, we performed a large metabolomic analysis in human brain in PD, taking into account cognitive subgroups and acute levodopa presence. As a result, we verified some known associations and discovered several new ones. We suggest that cognitive impairment in PD may be caused by two specific pathways, which rely on B vitamins and betaine as cofactors, the concentrations of which are modifiable by dietary supplementation. This is an important direction for future research.
You can find detailed results and their comprehensive discussion inside the article.
Follow the Topic
-
npj Parkinson's Disease
This journal publishes original basic science, translational and clinical research related to Parkinson's disease, including anatomy, etiology, genetics, cellular and molecular physiology, neurophysiology, epidemiology and therapeutic development and treatments.

Your space to connect: The Psychedelics Hub
A new Communities’ space to connect, collaborate, and explore research on Psychotherapy, Clinical Psychology, and Neuroscience!
Continue reading announcementRelated Collections
With Collections, you can get published faster and increase your visibility.
The neuroimmune-axis and ageing in Parkinson’s Disease
Publishing Model: Open Access
Deadline: Jul 15, 2026
Cognition - preclinical models, and preclinical unmet need
Publishing Model: Open Access
Deadline: Jul 27, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in