The basic science underlying drug design and discovery –
Published in Bioengineering & Biotechnology, Chemistry, and General & Internal Medicine

This month, in keeping with my traditions of writing and curiosity, I am interested in the design mechanisms underlying synthetic drug moieties and their analogues, to assess the capacity of these methods to identify a specific biological moiety for subsequent therapeutic interventions. It is possible to bottom-up engineer a drug candidate that is complementary in shape alongside its charge to a biological moiety by initially understanding the architecture of a biological target, to then examine their interaction dynamics, and binding properties for pharmacotherapy [Figure 1] [Zhou 2017].
This highly interdisciplinary research field seamlessly orchestrates medicinal chemistry, synthetic organic chemistry, biochemistry, molecular biology, cell biology, and mathematical/computational modeling to reveal the pharmacokinetics of novel drug candidates. These strategies can reveal the affinity, selectivity, and stability of preclinical drugs, for their eventual translational value from the bench to the clinic to treat diseases, to thus play a key role in the era of precision medicine [Mahapatra 2022].
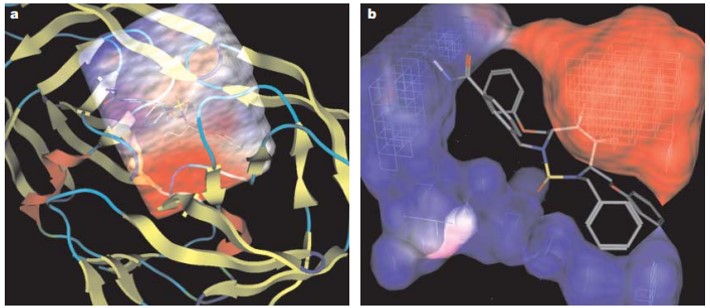
Figure 1: (a) Grid representations of an enzyme-substrate complex with an electrostatic potential around its active site in HIV protease. (b) And a close-up of the electrostatic potential grid of the enzyme around the bound inhibitor. Credit: [Kitchen 2004]
The art of rational drug design
At its origin, the process of drug discovery is based on the identification, characterization, and validation of a biological target. These preliminary efforts are followed by trouble-shooting assays with drug development and optimization, to identify the lead molecules. Suitable drug candidates can then be delineated to observe their absorption, distribution, metabolism, extraction, and pharmacokinetics. The final step in the drug discovery-to-development pipeline includes clinical trials to fit a bench to clinic framework [Tan 2015].
At the onset, synthetic chemists and structural biologists seek to minimize the process of trial-and-error associated with drug design and synthesis, to rationally design drugs that are structure-based and ligand-based moieties [Blundell 1996]. During rational drug design, scientists focus on target discovery, to screen a variety of analogues for their biochemical and structural properties and then study the interactions with a protein target, to precisely optimize drug-like candidates [Figure 2]. This usually involves the integration of synthetic organic chemistry methods such as nuclear magnetic resonance spectroscopy and X-ray crystallography, to support investigating the structural features of proteins [Khanna 2012].
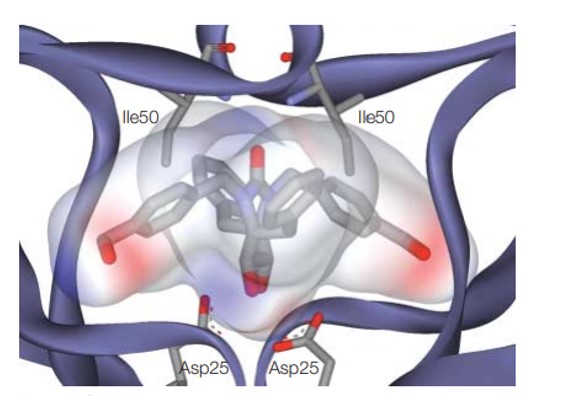
Figure 2: Docking and scoring in virtual screening for drug discovery: representing the electrostatic potential of a bound inhibitor. Credit: [Kitchen 2004]
On this note, Gertrude Elion Stein is a pioneering, Nobel prize-winning scientist who founded the strategy of rational drug design. Her efforts at drug discovery have forever altered and accelerated medical research to usher in a new era of antiviral therapy, including the first drug to treat AIDS – azidothymidine or AZT [Kresge 2008]. Using the rational approach, Elion further deduced that since all cells require nucleic acid to reproduce, both bacteria and tumors would require even more DNA to sustain their pace of growth.
The rational design of drugs inspired the process of precisely targeted DNA degradation by blocking its production via two competitive, synthetic compounds diaminopurine and thioguanine, which led to the elimination of several disease-causing pathogens, to ultimately also form a molecular therapy for cancer [Weber 2014]. This preliminary strategy of rational drug design forms the underlying basis of tailoring precision medicine efforts at present [Dugger 2017].
Core relationship – precision medicine: genomics and drug discovery.
Advanced technologies in genetic analysis and sequencing can reveal the relationship between genomics and drug development to provide a new window into the biology of disease, as seen with a patient undergoing genetic screening for the inherited disease epileptic encephalopathy [Figure 3]. This understanding of the physiological and molecular basis of disease is a key theme in precision medicine, which aims to transform drug development and clinical use. Biologists can develop preclinical models by analyzing human genomic variation to examine the underlying cause of disease in individual patients. Such efforts have found pioneering applications in oncology and in the move towards precision medicine as targeted immunotherapies to treat genetically inherited diseases [Dugger 2017].
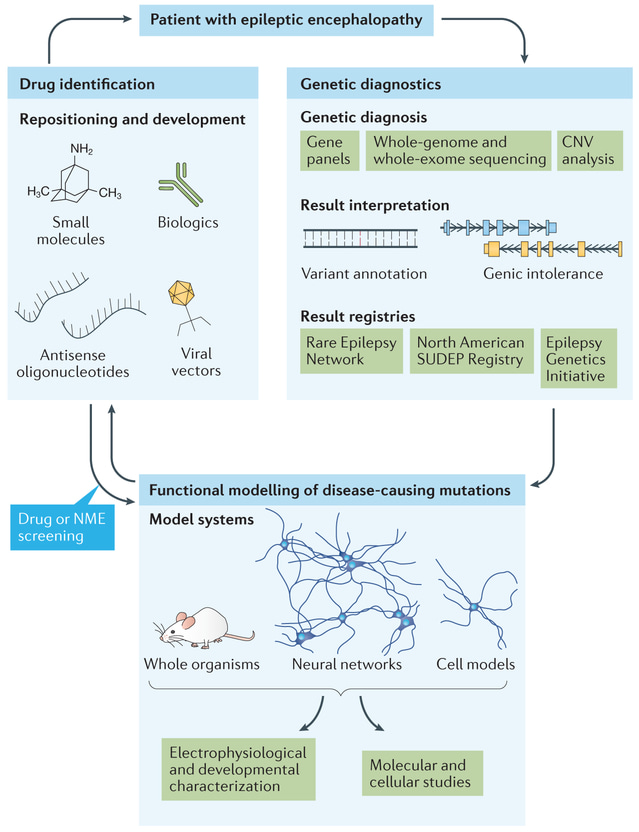
Figure 3: Precision medicine initiatives for highly genetic diseases – using epileptic encephalopathy as a model. Genetic testing of a patient with epileptic encephalopathy can include screening of an epilepsy gene panel or whole-exosome sequencing to detect single nucleotide variants or microarray analysis. Molecular and cellular studies can be conducted to assess protein-protein interactions with potential drug targets. Credit: [Dugger 2017].
Core theories – structure-based drug design.
The amalgamation of structural biology and synthetic chemistry is at the basis of structure-based drug design. The method identifies a three-dimensional biological structure of the target of interest, and its associated data via computational modeling to recognize proteins, receptors, and enzymes. It is possible to design high potent ligand molecules with a desired pharmacoepidemiologic character and a toxicity profile by identifying key steps of the method:
- Protein structure preparation
- Identifying the binding site of a protein of interest
- Ligand preparation
- Detecting the docking and scoring functionalities of the molecules
The core theory of drug design relies on the flexibility of the target through molecular docking and modeling efforts. This process, can, however, be limited by the non-static flexibility of the target throughout molecular docking and modeling processes, and the presence of water molecules that are cumbersome to the preparation process [Figure 2] [Kitchen 2004].
Drug molecules in synthetic medicinal chemistry typically maintain a stereochemistry or ‘chirality’ where the isomers or compounds with chiral similarity can be divided into constitutional or structural isomers, and stereoisomers [Akamatsu 2004]. Although enantiomers possess similar physicochemical properties, they express different pharmacological and biological activities due to the orientation of the substituents around the chiral center atom. As a result, clinically available drugs are typically dispersed in the racemate form relative to pharmacokinetic and pharmacodynamic attributes.
A guide to quantifying structure-activity relationships in pharmacology
The structure-activity relationship of a drug molecule relative to its biological activity can be defined by quantifying the relationship between the chemical structure or the pharmacophore relative to a biological moiety. Biochemists can accomplish this by implementing a small library of molecules to exhibit the desired therapeutic activity, where associations between the groups can reveal their pharmacological potential [Mahapatra 2022].
It is possible to initially quantify the structure-activity relationship of ligands by using experimental and biological measurements chosen from diverse chemical libraries. Researchers can then establish chemical descriptors that accompany diverse structural and physicochemical properties of such drug candidates to investigate associations between molecular descriptors and their combined pharmacological activity. For instance, the mathematical associations between molecular descriptors and the efficiency of pharmacological structures are integral to identify the quantitative structure-activity relationship of pharmaceutical drug compounds [Stella 2007].
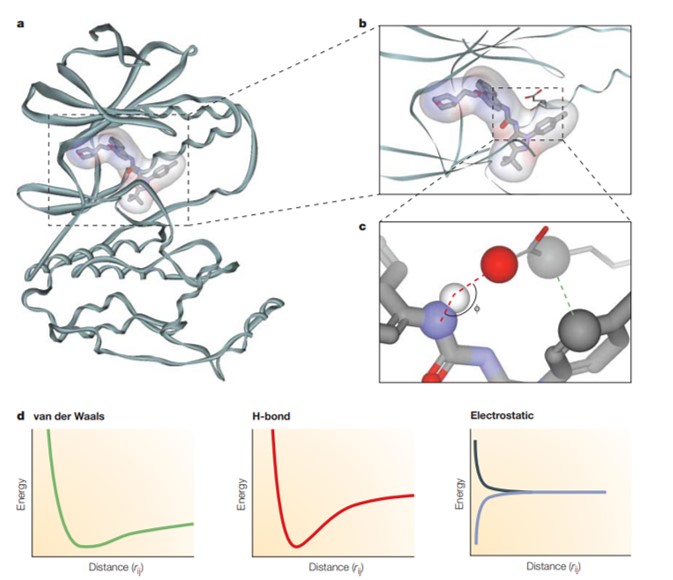
In 1926, Scientist A. J. Clark revealed the mathematical association between receptors and ligands to explain the interaction of oxygen and carbon dioxide with hemoglobin [Clark 1926]. Indeed, the binding affinity between ligands and the active site of a receptor can be determined with mathematical precision to estimate the total potential energy of a system, also known as a force field. Such interactions are contained as bonded interactions or non-bonded interactions to include van der Waals forces, H-bonding, and electrostatic forces for the latter [Figure 4]. The force-field equation relies on classical physics or molecular mechanics, but it is not suited for complex interactions that involve the making or breaking of covalent bonds between the ligand and receptor.
Pharmacokinetics and pharmacodynamics
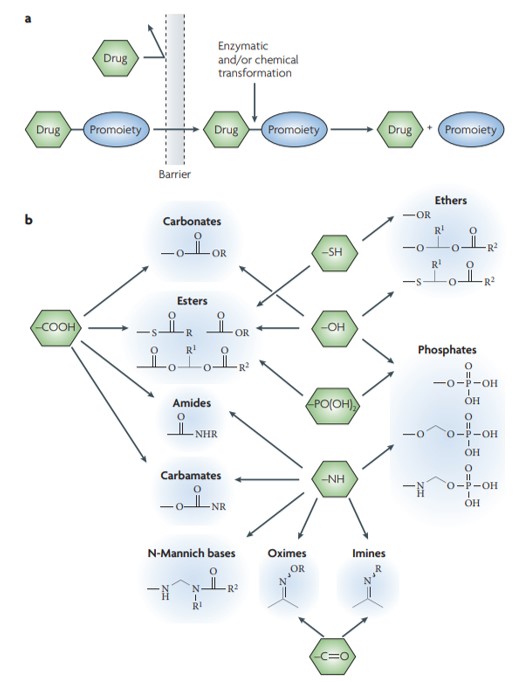
Figure 5: A simplified representative illustration of the concept of prodrugs. (a) The prodrug is typically pharmacologically inactive, linked to its parent compound via non-toxic, bio-reversible groups that are enzymatically labile. b) Common functional groups on parent drugs that are amenable to prodrug design, where most prodrug approaches require a synthetic handle on the drug. Credit: [Rautio 2008].
The field of drug discovery is more aptly defined by pharmacokinetics and pharmacodynamics; the branches of pharmacology that describe the impact of drugs on the body [Mahapatra 2022]. These include interactions between drug molecules and their target receptors/enzymes, while pharmacokinetics mainly define the therapeutic impact and the profile of a drug candidate both in vitro and in vivo.
Aspects of pharmacokinetics include understanding the dynamics of drug metabolism and of prodrugs or bio-inactive compounds that can undergo chemical modification or enzymatic catalysis, to either form or release the parent drug/active candidate to treat a disease. This strategy is quite popular in drug discovery and development to overcome the challenging kinetics associated and ensure the suitability of drug molecules [Figure 5] [Rautio 2008].
Since prodrugs can influence the efficacy, toxicity and distribution of the parent molecule, a range of parameters are taken into consideration to design the compounds. Each compound has a variety of functional molecules from carbonyl, hydroxyl, carboxylic acid, amine, and phosphate as well as phosphonate. These drugs are defined by oral absorption improvement, enhanced aqueous solubility and the mechanisms of sustained drug activity [Golan 2011].
Designing drugs to attenuate a pathological pathway.
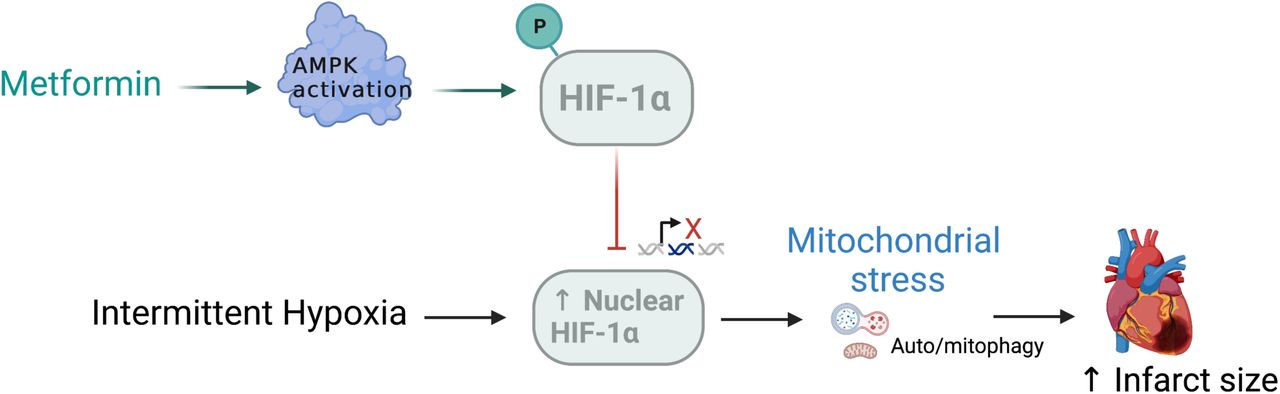
All methods of drug design and discovery are shaped to attenuate or hinder a biomechanism in a pathological pathway. For instance, the commercial drug Metformin or N, N-Dimethylimidodicarbonimidic diamide is an antidiabetic drug that can alter the hypoxic HIF-1α pathway by influencing the adenosine-monophosphate activated protein kinase (AMPKα2), with broad impact to treat a variety of diseases including cardiac stress, ischemia, and potentially renal biomineralization; diseases that are all commonly driven at the onset by oxidative stress [Moulin 2024, Shalmashi 2008].
New findings in mathematical and computational biology in medicine also highlight the capacity of high concentrations of metformin to reduce oxidative stress, injury, and inhibit the growth and migration of renal carcinoma cells [Figure 6] [Liu 2022]. Metformin can reverse the death of cardiomyocytes that are induced by intermittent hypoxia and mitophagy, by decreasing the nuclear expression of the hypoxia-inducible factor 1α (HIF-1α); a key regulator of the hypoxia/ischemia-reperfusion pathway [Figure 7].
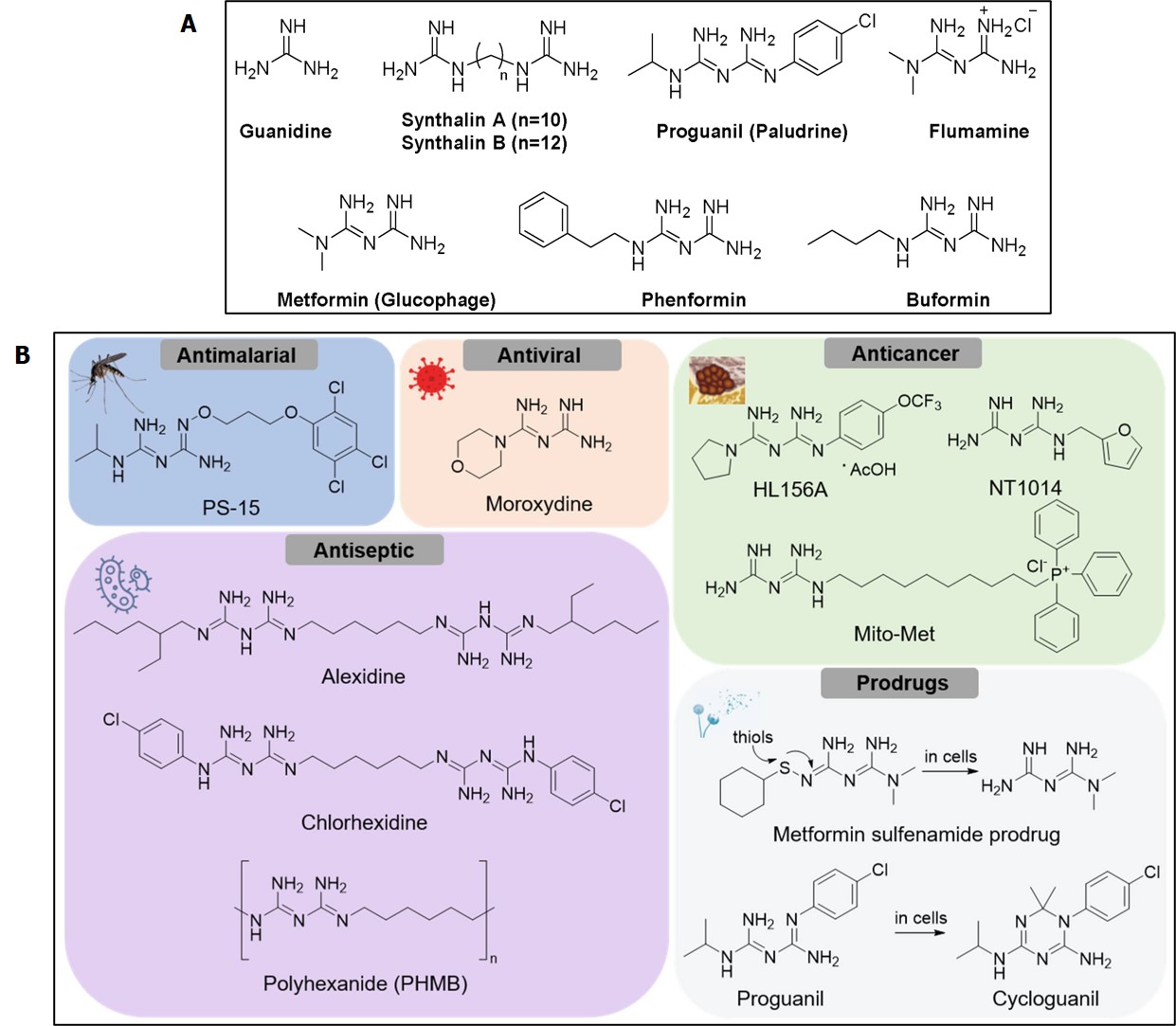
Figure 6: A) chemical structures of metformin and structurally similar biguanides. B) chemical structures of metformin analogues with diverse biological activities. Credit: [Rusanov 2022].
The drug also eliminated the decrease of P62 expression (associated with autophagy) that resulted from intermittent hypoxia to play a crucial role during pathological cellular processes such as DNA damage response, ageing, chronic inflammation, and carcinogenesis. Since the metformin compound can rescue mitochondria from undergoing mitophagy under hypoxic conditions and protect hypoxic cardiac tissue and other organs against ischemia-reperfusion, the outcomes highlight the influence of exploring the drug and its analogues across the spectrum of oxidative-stress related disease origins [Rusanov 2022].
Outlook
This article is simply an overview of the biochemical basis of rational drug design and discovery, with input from the interdisciplinary research fields of molecular biology and computational methods to facilitate a bench-to-clinic pharmacoepidemiology framework. I bring together a few core theories and core relationships to explore the complexity of the process and highlight the legacy of Gertrude Elion as a pioneer of the rational drug design process. Drug discovery is a complex, multistage, and versatile method with substantial capacity to advance and vary with newer technologies and design paradigms. So, this is simply a starter post to the theme, with capacity to expand on the context.
The avenues of drug discovery can benefit from computer-aided drug design, equally in the fields of medicinal chemistry and genomics-based precision medicine to allow researchers to bypass/hasten multiple procedures in the pipeline to screen potential clinical candidates in a short timeframe for FDA approval. The process of drug discovery includes multistage optimizations for cost-effective findings to advance the field of medicine and healthcare.
Header Image: A schematic representation of revolutionizing drug discovery via the Drug Target Review.
References
- Zhou S. et al. 2017, Drug design and discovery: Principles and Applications, Molecules, MDPI
- Mahapatra M. et al. 2022, Fundamental considerations in drug design, Elsevier
- Tan S. et al. 2015, Alexander Fleming, Discoverer of Penicillin, Singapore Medical Journal
- Blundell T. et al. 1996, Structure-based drug design, Nature
- Khanna I. et al. 2012, Drug discovery in pharmaceutical industry: productivity challenges and trends, Drug Discovery Today.
- Kresger N. et al. Developing the Purine Nucleoside Analogue Acyclovir: the Work of Gertrude B. Elion, The Journal of Biological Chemistry.
- Weber G et al. 2014, DNA damaging drugs, Molecular Therapies of Cancer.
- Kitchen D. et al. 2004, Docking and scoring in virtual screening for drug discovery: methods and applications, Nature Reviews Drug Discovery
- Bacilieri M. et al., 2006, Ligand-based drug design methodologies in drug discovery process: an overview, Current Drug Discovery Technologies.
- Chang C. et al. 2006, Pharmacophore-based discovery of ligands for drug transporters, Advanced Drug Delivery Reviews.
- Akamatsu M. et al. 2004, Current state and perspectives of 3D-QSAR, Current Topics in Medicinal Chemistry.
- Stella V. et al. 2007, Prodrug strategies to overcome poor water solubility, Advanced Drug Delivery Reviews
- Clark A. J. et al. 1926, The antagonism of acetyl choline by atropine, The Journal of Physiology.
- Rautio J. et al. 2008, Prodrugs: design and clinical applications, Nature Reviews Drug Discovery.
- Moulin S. et al. 2024, Metformin protects the heart against chronic intermittent hypoxia through AMPK-dependent phosphorylation of HIF-1α, BioRxiv
- Shalmashi A. 2008, New Route to Metformin Hydrochloride (N,N-dimethylimidodicarbonimidic diamide hydrochloride) Synthesis, Molbank, MDPI
- Liu Y. et al. 2022, High-Concentration metformin reduces oxidative stress injury and inhibits the growth and migration of clear cell renal cell carcinoma, Computational and Mathematical models in medicine.
- Rusanov D. et al. 2022, Biological properties of transition metal complexes with metformin and its analogues, Pharmaceuticals, MDPI
I am an interdisciplinary researcher with a strong commitment to bioengineering, biochemistry, organ-chips and molecular biology. As well as biomechanics and biomineralization in the broader context of medicine. I completed my PhD at the University of Sydney Australia in December 2016, and travel often, find me on Twitter and irl.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in