The IL-3, IL-5 and GM-CSF Common Receptor Beta Chain Mediates Oncogenic Activity of FLT3-ITD Positive AML
Published in Cancer
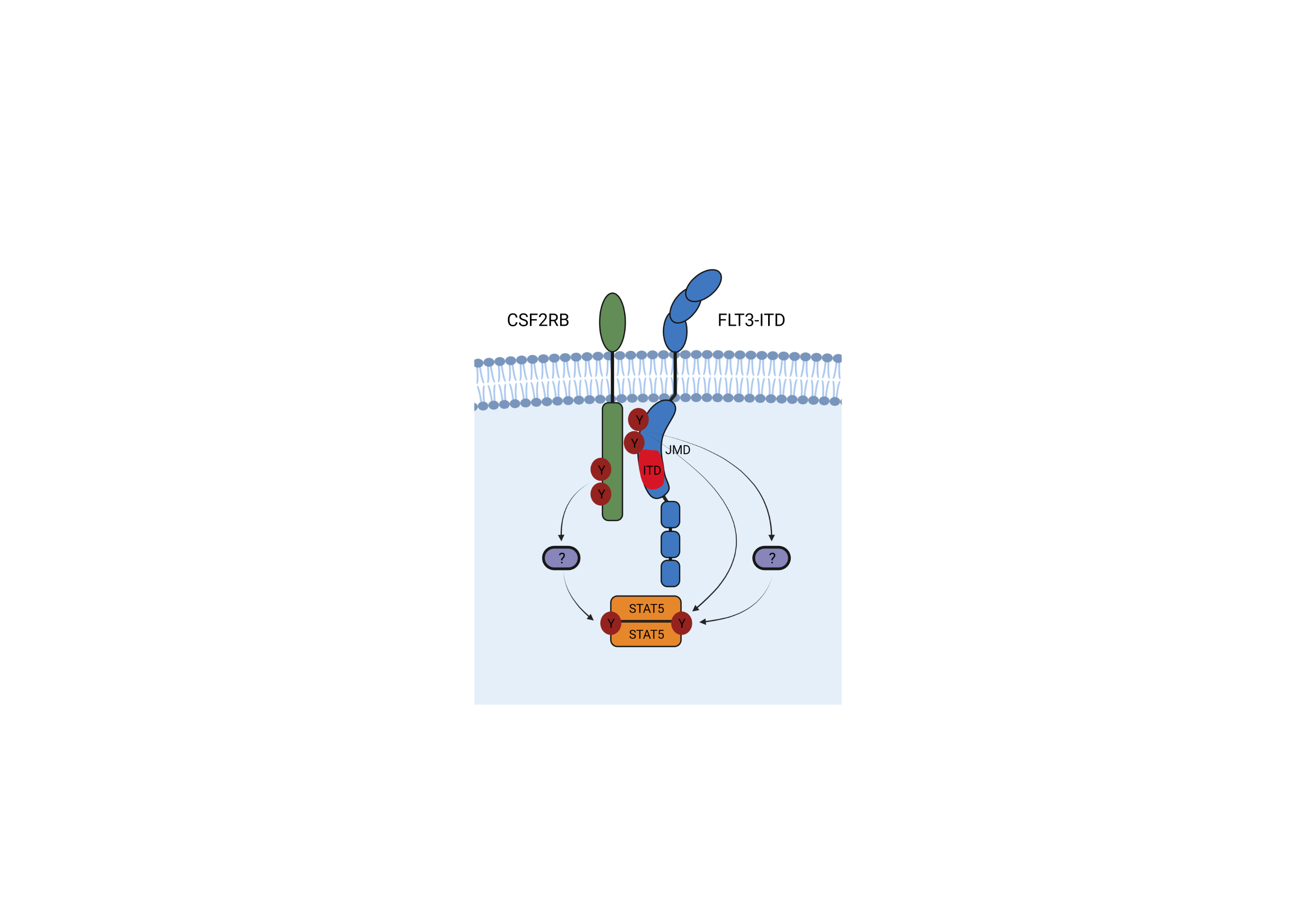
FLT3-ITD is the most predominant mutation in AML being expressed in about one third of AML patients and is associated with a poor prognosis. Many questions regarding its oncogenic potential and signalling still remain unanswered. In our recently published work, we were able to identify JAK-mutations as resistance mechanism against FLT3 tyrosine kinase inhibitors (TKIs) that activate the CSF2RB – STAT5 signalling pathway and reinstate oncogenic signaling that way (1). Additionally we were able to demonstrate that CSF2RB is also activated in uninhibited FLT3-ITD positive leukemic cell lines. Usually, CSF2RB, the common beta chain of IL-3, IL-5 and GM-CSF receptors (CD131, IL3RB), becomes activated after the respective alpha chaines bound the respective ligands and assembled a receptor complex that activates associated JAKs and ultimately leads to STAT5 activation (2,3). Strikingly, in FLT3 TKI-naïve JAK wild type (WT) cells, CSF2RB was phosphorylated in a JAK2 independent, but FLT3-ITD dependent fashion that disappeared upon FLT3 inhibition, suggesting that CSF2RB participates in FLT3-ITD downstream signal activation. The aim of this work was therefore to investigate the role of CSF2RB in FLT3-ITD positive AML.
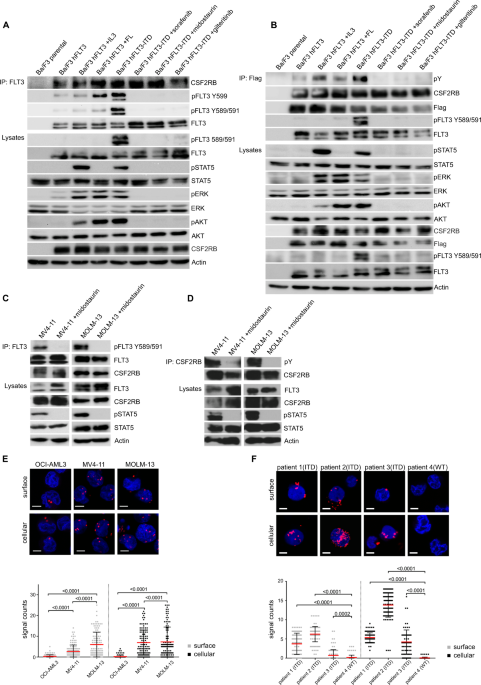
First, we could demonstrate that FLT3-ITD directly interacts with CSF2RB in Ba/F3 cells expressing FLT3-ITD as well as in FLT3-ITD positive primary AML cell lines MOLM-13 and MV4-11 via co-immunoprecipitation. We could also dectect the interaction using proximity ligation assays in the above mentioned cell-lines as well as in blasts isolated from AML patients. CSF2RB was phosphorylated in this context and this phosphorylation was abolished after treatment with different FLT3-TKIs whereas the interaction between CSF2RB and FLT3-ITD remained unaltered. CSF2RB was only activated when the tyrosines 589 and 591 of FLT3 were phosphorylated which was not the case upon stimulation of FLT3 wild type with FLT-ligand. The same was determined for downstream sinalling via STAT5.
To elaborate the significance of activated CSF2RB in FLT3-ITD positive AML, we performed a series of knockdown experiments. CSF2RB knockdown impaired proliferation in all FLT3-ITD expressing cell lines and sensitized them to growth inhibition induced by the FLT3-inhibitor midostaurin. Small concentrations of midostaurin synergized with CSF2RB knockdown to block the outgrowth of our knockdown cell lines. Usage of the more FLT3 specific inhibitor gilteritinib confirmed these results. Furthermore, CSF2RB knockdown decreased the phosphorylation of STAT5 in MOLM-13 and MV4-11 cells. We next transplanted CSF2RB knockdown MOLM-13 cells in a xenograft model. Expression of a luciferase reporter served for visualization of leukemic cell expansion after engraftment. Consistent with our in vitro findings, CSF2RB knockdown cells showed significantly less expansion in vivo and the mice had a longer survival compared to controls. Using the same cells in a subcutaneous tumor model, CSF2RB knockdown tumors showed slower growth rates and almost absent phosphorylation of STAT5. And finally, bone marrow from CSF2RB deficient mice transfected with FLT3-ITD displayed decreased colony formation capacity and delayed disease onset together with increased survival upon transplantation into lethally irradiated mice. Together, these experiments demonstrate the biologic relevance of CSF2RB in mediating FLT3-ITD dependent transforming capacity and oncogenic potential in vitro and in vivo.
To further elaborate the interaction between CSF2RB and FLT3-ITD, we identified that the interaction of the two receptors occurs in the juxtamembrane domain (JMD) using a GST pulldown mapping approach. Next we assessed that FLT3-ITD dependent CSF2RB phosphorylation required phosphorylation of the FLT3 juxtamembrane domain at tyrosines 589 or 591. Both tyrosines are repeated in the majority of known ITD sequences, are autophosphorylated in FLT3-ITD, not phosphorylated upon FLT-ligand stimulation of FLT3 WT and are required for FLT3-ITD mediated phosphorylation of STAT5. Expression of differentially mutated tyrosines within the JMD of FLT3-ITD identified that the phosphorylation of CSF2RB by FLT3-ITD requires only one of the tyrosines 589 and 591 to be phosphorylated. Additional repeated tyrosines within the duplicated sequence are not required. Expressing a variety of known FLT3-ITDs and alanine mutants thereof in Ba/F3 cells indicated that the length, sequence and insertion site of the ITDs are not critical for FLT3-ITD dependent CSF2RB phosphorylation, STAT5 activation and transformation.
We here show for the first time that CSF2RB directly interacts with FLT3 and is phosphorylated in a FLT3-ITD dependent fashion. It participates in FLT3-ITD induced activation of STAT5 and plays a pivotal role in promoting the oncogenic potential of FLT3-ITD in vitro and in vivo, suggesting it to be a rational drug target in FLT3-ITD positive AML. Our data add another piece to the puzzle of how FLT3-ITD activates STAT5. Presumably, interaction between cytokine receptors and mutant receptor tyrosine kinases like FLT3-ITD might represent an underestimated mechanism of aberrant oncogenic signaling.
References
- Rummelt C, Gorantla SP, Meggendorfer M, Charlet A, Endres C, Döhner K, et al. Activating JAK-mutations confer resistance to FLT3 kinase inhibitors in FLT3-ITD positive AML in vitro and in vivo. Leukemia. 2020 Nov 4;
- Mui AL, Wakao H, O’Farrell AM, Harada N, Miyajima A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 1995 Mar 15;14(6):1166–75.
- Hercus TR, Dhagat U, Kan WLT, Broughton SE, Nero TL, Perugini M, et al. Signalling by the βc family of cytokines. Cytokine Growth Factor Rev. 2013 Jun 1;24(3):189–201.
Follow the Topic
-
Leukemia
This journal publishes high quality, peer reviewed research that covers all aspects of the research and treatment of leukemia and allied diseases. Topics of interest include oncogenes, growth factors, stem cells, leukemia genomics, cell cycle, signal transduction and molecular targets for therapy.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in