
Imagine you have a world map in front of you. Now pick up a coin and throw it randomly on the map, and repeat it, time and time again. It sounds boring, but maybe we can find something interesting. OK, here comes some questions: how often do your coin overlap with the ocean part or the land part of the map? If the coin lies totally on the land part, how often do your coin covers a whole country, lies within one country, or overlap with two or even more countries (Figure 1)?
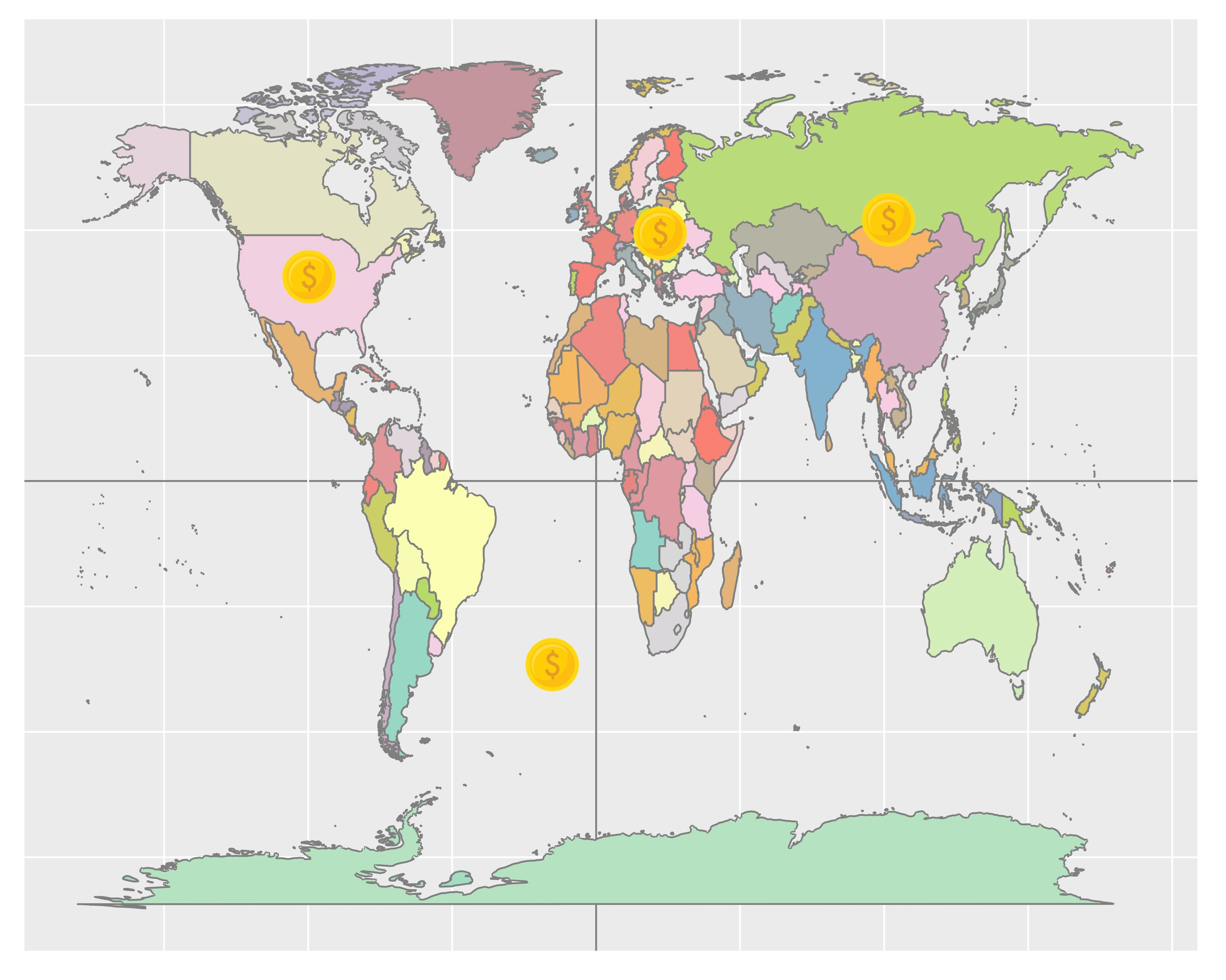
Figure 1. Illustration of throwing coin randomly on a world map.
Our story started from a similar perspective. As illustrated in the title, we seek to investigate the landscape of youngest duplicates (polymorphic or only present in some individuals) in fly and human. Duplication is one of the most important mechanisms underlying evolutionary novelty; it ranges from small scale duplication to whole genome duplication, brings in new genetic material to the genome, and thus make the emergence of new function possible during evolution. The object of our paper is small scale tandem duplication, which is 50 bp to 25 kb long and located adjacent to its parental sequence. Such duplication is relatively small compared with the whole genome, and it may overlap with intergenic or genic region, just like a coin may overlap with the ocean or land part of a world map. So, our first task was to describe the landscape of duplication: in the very beginning phase of duplication, i.e., among these polymorphic duplicates, how many genes are duplicated? Moreover, how many genes are completely or partially duplicated, or just duplicated in the intron (Figure 2)?
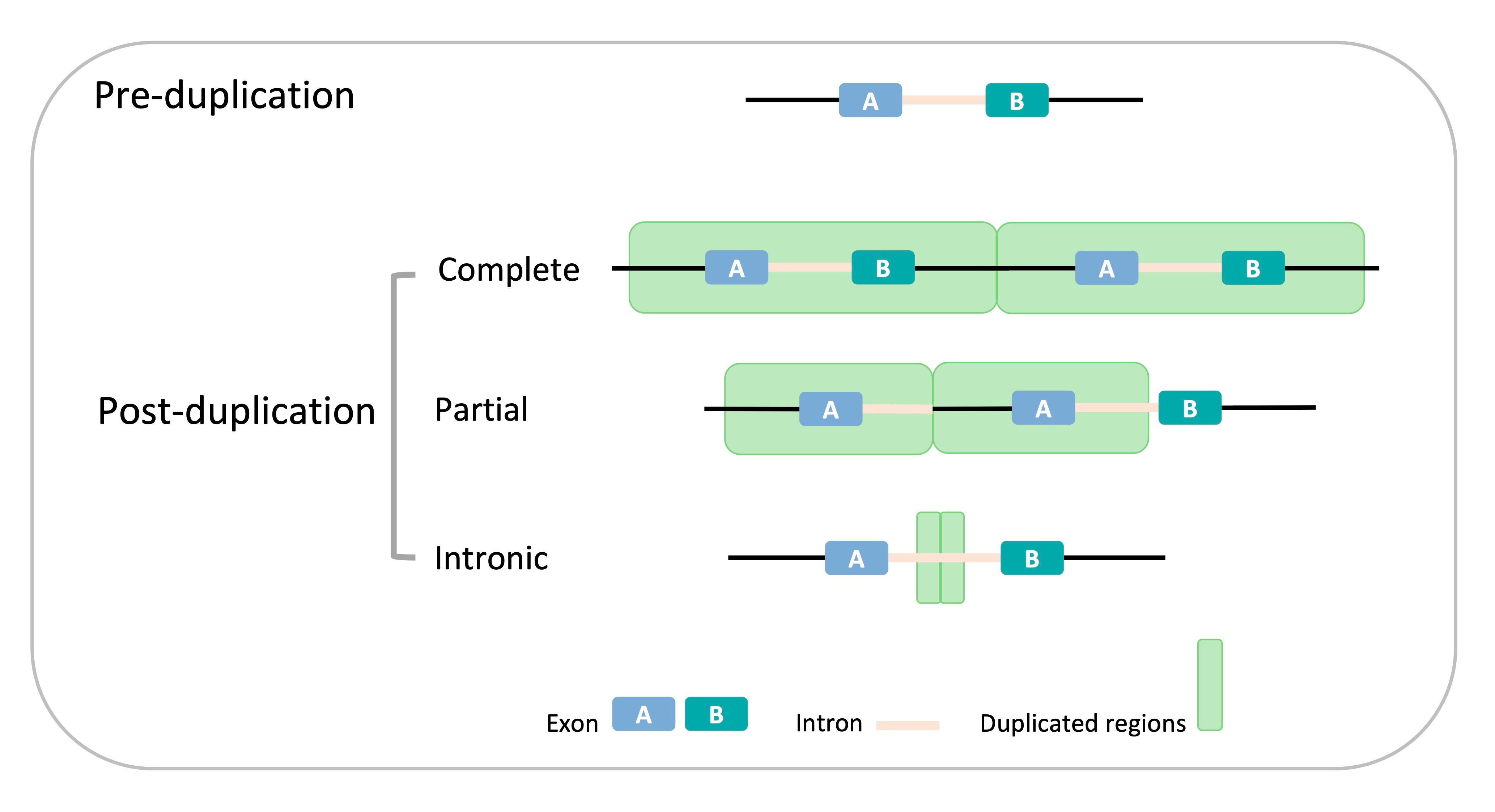
Figure 2. Schematic classification of duplicate genes.
We found 270 duplicates in Drosophila and 964 duplicates in humans and classified them into complete duplicates, partial duplicates and intronic duplicates. Interestingly, only a small fraction of genes is completely duplicated in both human and fly. Besides, fly harbors a higher proportion of partial duplicates and a lower proportion of intronic duplicates than humans. This discrepancy could be explained by two factors: genomic architecture and duplication size. To take the genomic architecture as an example, fly genome is much denser than human genome; introns and exons contribute respectively to 17% and 20% of the fly genome, but to 40% and 1% of the human genome. Just like throwing a coin on the world map, if the coin stop roaming and lies in Europe, it usually partially overlaps with two or more countries; but if it stops in north America, it is highly possible that the whole coin is lying within US or Canada. So, this is our first observation.
We have observed an overall landscape of polymorphic duplicate. We then take steps forward. Why is the complete duplicate so rare in both fly and human? The answer may come from dosage constraint. Different proteins in organism usually function as a molecular biological machine, which is called a protein complex. The amount of different proteins in a protein complex should follow the principle of stoichiometric balance, so the dosage increase of gene that encodes a component of a stable protein complex may disrupt the stoichiometric balance and thus be toxic to the organism. In line with this hypothesis, we found that complete duplicates tend to show dosage increase and members of protein complexes are depleted among complete duplicates when compared to the genomic background. Therefore, in summary, complete duplicates are a minority of all duplicates, show dosage increases and are subject to dosage constraints imposed by stoichiometry in protein complexes.
Maybe you still remember that we found a lot of partial duplicates. So, what’s happening to them? The story of partial duplicates is more complex than complete ones where the main player is no longer dosage but exon shuffling. Since a gene generally comprises several parts, e.g., introns and exons, post-duplication changes in gene structure should depend on the boundaries of duplications (Figure 2). Obviously, genome architecture also works on partial duplicates: duplications potentially leading to gene fusions are more frequent in flies than in humans, which is consistent with the higher gene density in flies. In contrast, humans showed a marginal enrichment of internal duplications compared with flies, likely due to the greater gene size relative to duplication size found in humans. Moreover, these partial duplicates bring in some structural changes and generate new forms of pre-existing gene. For example, shuffling often uses intronic breakpoints and generates in-frame proteins in humans. Take the C6 gene as an instance (Figure 3), two internal exons were duplicated and the post-duplication protein have three possible forms by alternative splicing. Thus, here alternative splicing could occur to rescue the pre-duplication ORFs, while still keep the possibility of generating a new protein. This mechanism is quite a nice example of the canonical ‘gene in pieces’ theory proposed by Gilbert.
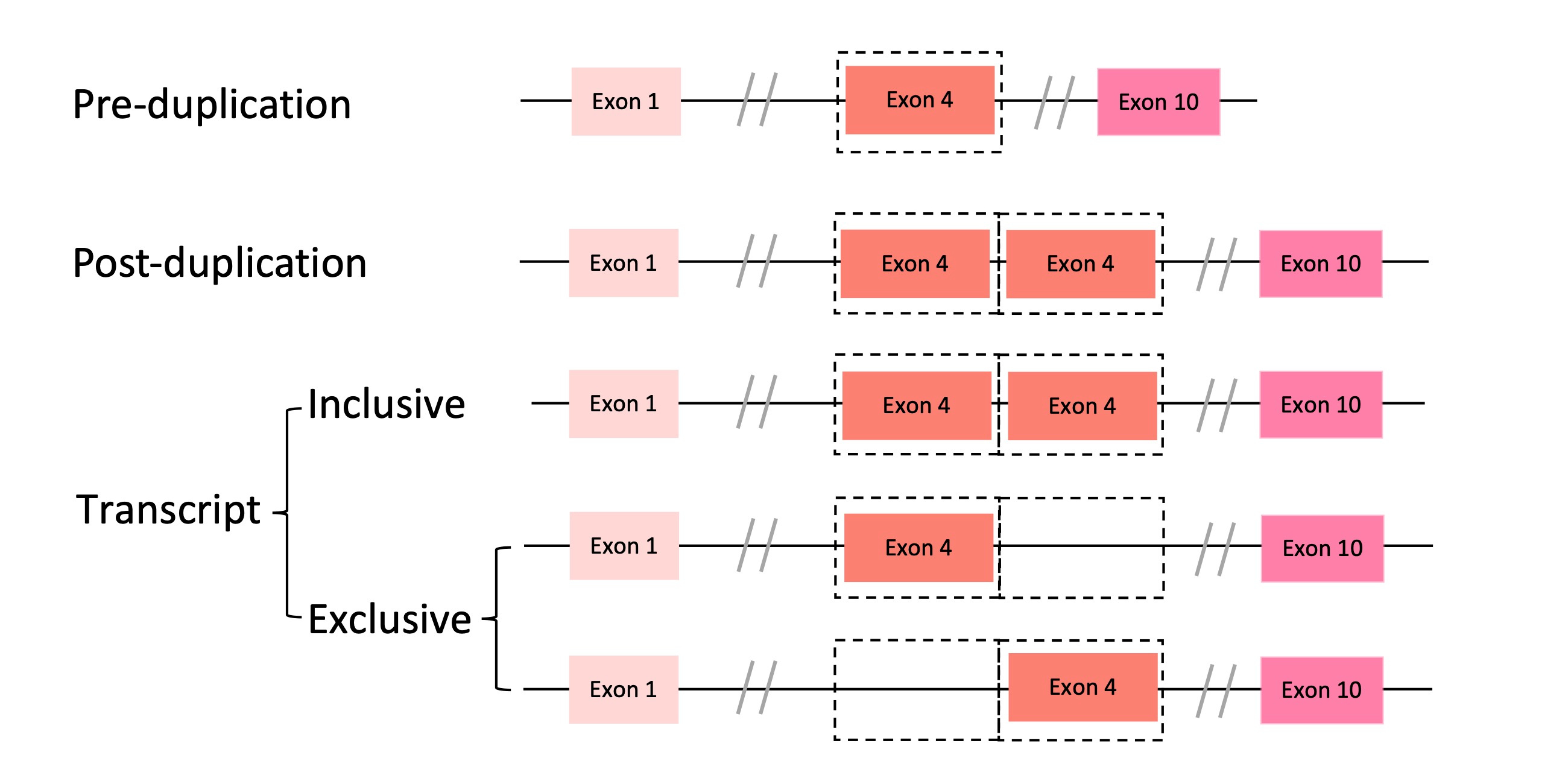
Figure 3 Alternative isoforms of C6.
Our introduction to the paper may stop here, but for the scientific question, ‘now this is not the end... But it is, perhaps, the end of the beginning’. Here we would like to talk a little bit more about the concept of variation. In recent decades we are familiar and work with different types of variation, e.g., SNP or duplications. But that’s not enough. Perhaps that may even not be the right way. In our opinion, the final portrait of variation should be in a T-2-T (telomere-to-telomere) way: T-2-T assembly of every chromosome from different individuals, T-2-T alignment of these chromosomes, and finally T-2-T portrait of all variations, from 1-bp point mutations to whole chromosome duplication. Now we are standing on the door of such a T-2-T palace: recently human X chromosome and Arabidopsis thaliana chr3 and chr5 are assembled successfully in a T-2-T way. Such advances in methodology must change the way we think about variation, trigger new attempts to classify and record all variations in any given genetic pool, and motivate researchers to propose new analysis tools and methods to exploit these T-2-T genetic variation data. Under such a new framework of T-2-T variation, our current observation of polymorphic duplicates may be no more than naive, and we would expect to see, or to contribute to, new sights of the landscape of all genetic variation, the consequence of gene in pieces under the T-2-T point of view, and genome evolution.
Read the new paper at https://www.nature.com/articles/s41559-021-01614-w
Application of state-of-the-art sequencing technology and methodology in Chinese traditional medicinal research.
Follow the Topic
-
Nature Ecology & Evolution
This journal is interested in the full spectrum of ecological and evolutionary biology, encompassing approaches at the molecular, organismal, population, community and ecosystem levels, as well as relevant parts of the social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Understanding species redistributions under global climate change
Publishing Model: Hybrid
Deadline: Dec 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in