The Lock and Key of Puerperal Sepsis
Published in Microbiology

Puerperal sepsis is a life-threatening disease that affects women within the first three days of childbirth. In the early 19th Century, maternity wards witnessed devastating outbreaks of severe puerperal sepsis, also known as childbed fever or puerperal fever, with a fatality rate as high as 70 to 80 per cent (Colebrook 1956). Ground-breaking research by Ignaz Semmelweis demonstrated that puerperal sepsis was preventable through the introduction of hand hygiene in maternity wards. Since then, researchers have been trying to identify both the causative microorganism and the mechanism by which the microorganism causes puerperal sepsis. Louis Pasteur and others found that puerperal sepsis was caused by streptococci, and the species involved was later identified as Streptococcus pyogenes, also known as Group A Streptococcus (GAS) (Lancefield and Hare 1935). Although S. pyogenes is a single bacterial species, its global population exhibits considerable genetic diversity, reflected in variations in surface proteins and interactions with the human host. Molecular studies of S. pyogenes have repeatedly associated a surface protein called R28 puerperal sepsis with isolates that caused puerperal sepsis outbreaks (Colman et al. 1993).
The hallmark of puerperal sepsis is the infiltration of S. pyogenes from the female reproductive tract into the bloodstream. However, this occurrence is not frequent in women unless they recently gave birth. It is likely that wounds in the epithelia that are generated during childbirth, therefore, provide a portal of entry for S. pyogenes to cause puerperal sepsis.
How could a single bacterial protein, R28, promote the capacity of S. pyogenes to cause puerperal sepsis outbreaks? One explanation was that R28 acts as a “key” that unlocks the defence mechanisms of host cells by interacting with a receptor - or “lock” - on the surface of human cells. As such, the lock-and-key interaction would open the door for S. pyogenes to overcome host defences, establish infection and cause puerperal sepsis.
Our new study shows that R28 specifically targets a “lock” called CEACAM1 that is expressed on the surface of many different human cell types. Since CEACAM1 is an immunomodulatory receptor, forming this lock-and-key interaction provides S. pyogenes with a mechanism to overcome many different defence mechanisms and the opportunity to cause invasive infections and puerperal sepsis.
We previously demonstrated that a related bacterium called Streptococcus agalactiae (also known as Group B Streptococcus) interacts with CEACAM1 through the expression of a “key” on its surface called β protein (van Sorge et al. 2021). More specially, this interaction was formed by a domain in β protein - or “groove” on the key - called IgI3. Since the R28 protein possesses a similar domain, we were led to hypothesize that CEACAM1 is the cognate target “lock” of R28. We found that R28 directly binds to human CEACAM1 with high specificity using biochemical analyses and solved the mechanism by which the interaction is formed through structural analyses. In parallel, we uncovered that R28-expressing S. pyogenes interact with CEACAM1 expressed on the surface of human cells.
Bacterial attachment to epithelial barriers is the first crucial step in bacterial infection. This often occurs when bacterial “keys” bind to host “locks” that then attach the bacterium to the host cell. A previous study has shown that R28 promotes bacterial attachment to human cervical epithelial cells (Stålhammar-Carlemalm et al. 1999). In our research, blocking the availability of CEACAM1 was sufficient to reduce the attachment of R28-expressing S. pyogenes to human cervical epithelial cells. CEACAM1 is known to regulate epithelial cell division and cell migration (Gray-Owen and Blumberg 2006; LeBlanc et al. 2011). This led us to investigate whether this lock-and-key interaction disrupted the repair of wounds in epithelial cell layers. R28 significantly delayed the repair of wounds in human cervical epithelial cells, but this was prevented when interaction with CEACAM1 was blocked. Since CEACAM1 is expressed on the outer surface of epithelial cells lining the female reproductive tract, we believe our results mean that R28 engagement with CEACAM1 equips S. pyogenes with a key mechanism to prolong the time it has to invade wounded tissues following childbirth.
Overcoming attacks from the immune system is also required for a bacterium to cause invasive infections in the host. Indeed, S. pyogenes is known to have an arsenal of lock-and-key mechanisms that help to suppress the immune response and promote bacterial survival (Laabei and Ermert 2019; Barnett et al. 2015). CEACAM1 is also an immunomodulatory receptor expressed in a variety of immune cells resident in our tissue and circulating in our blood, including neutrophils, monocytes, T cells and B cells (Gray-Owen and Blumberg 2006). This led us to test whether R28 and CEACAM1 interactions help S. pyogenes to disrupt human immune responses and escape immune killing. We infected human cervical tissue sections with S. pyogenes strains and found that the expression of R28 disrupted the production of pro-inflammatory cytokines and chemokines. Having characterized that R28-expression disrupts the immune response in tissue, we then evaluated its significance in evading the killing by immune cells of the blood. We found that the expression of R28 reduced the killing of S. pyogenes by neutrophils and in blood, but also that this was prevented when interaction with CEACAM1 was blocked. These results suggest that S. pyogenes uses R28 as a "key" to unlock the functions of innate immune cells by binding to the "lock" of CEACAM1. This would facilitate S. pyogenes to replicate within the bloodstream and cause puerperal sepsis.
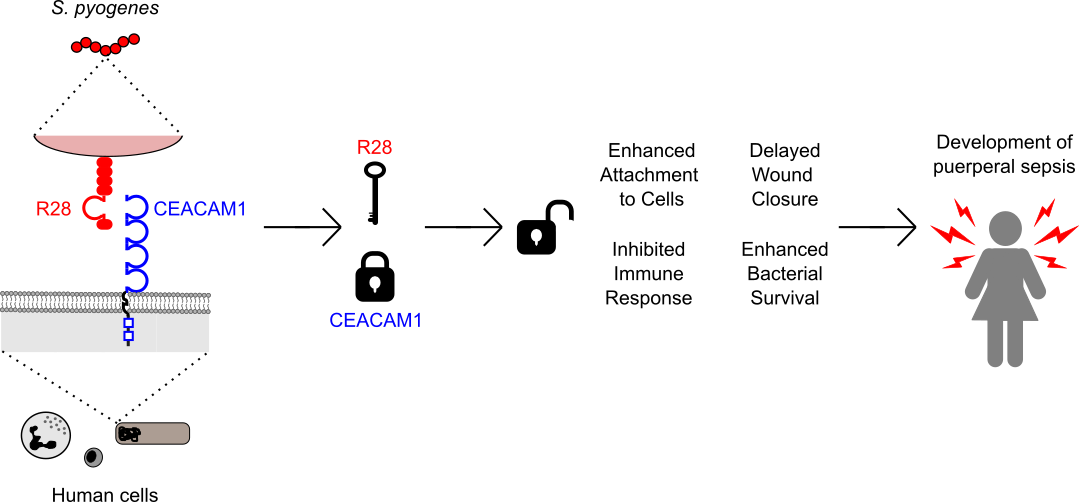
Schematic of the interaction between the S. pyogenes R28 protein and the human cognate receptor CEACAM1. This lock-and-key unlocks the defence mechanisms of host cells that lead to events that would promote the development of puerperal sepsis in women shortly after childbirth.
Puerperal sepsis has had an instrumental role in the development of germ theory, hand hygiene, antiseptic techniques, and molecular epidemiology. It is considered a classic example of a hospital-acquired infection. We feel our work presents a new chapter in puerperal sepsis research that may help to develop strategies to prevent this disease. Our findings demonstrate that the key bacterial molecule, R28, opens many doors to promote S. pyogenes pathogenesis by targeting a single lock molecule, CEACAM1, that is expressed in a variety of human cell types. This challenges the conventional belief in molecular microbiology that a single lock-and-key interaction is only relevant for overcoming a single host defence mechanism on the path to causing invasive disease.
To see the full story, check out the publication in Nature Communications:
https://www.nature.com/articles/s41467-023-37732-1
References
Barnett T.C., Cole J.N., Rivera-Hernandez T., Henningham A., Paton J.C., Nizet V., and Walker M.J. 2015. “Streptococcal Toxins: Role in Pathogenesis and Disease.” Cellular Microbiology 17 (12): 1721–41. doi: 10.1111/cmi.12531
Colebrook L. 1956. “The Story of Puerperal Fever; 1800 to 1950.” British Medical Journal 1 (4961): 247–52. doi: 10.1136/bmj.1.4961.247
Colman G., Tanna A., Efstratiou A., and Gaworzewska E.T. 1993. “The Serotypes of Streptococcus Pyogenes Present in Britain during 1980-1990 and Their Association with Disease.” Journal of Medical Microbiology 39 (3): 165–78. doi: 10.1099/00222615-39-3-165
Gray-Owen S.D. and Blumberg R.S. 2006. “CEACAM1: Contact-Dependent Control of Immunity.” Nature Reviews. Immunology 6 (6): 433–46. doi: 10.1038/nri1864
Laabei M. and Ermert D. 2019. “Catch Me If You Can: Streptococcus Pyogenes Complement Evasion Strategies.” Journal of Innate Immunity 11 (1): 3–12. doi: 10.1159/000492944
Lancefield R.C. and Hare R. 1935. “The Serological Differentiation of Pathogenic and Non-Pathogenic Strains of Hemolytic Streptococci from Parturient Women.” The Journal of Experimental Medicine 61 (3): 335–49. doi: 10.1084/jem.61.3.335
Lancefield R.C. and Perlmann G.E. 1952. “Preparation and Properties of a Protein (R Antigen) Occurring in Streptococci of Group A, Type 28 and in Certain Streptococci of Other Serological Groups.” The Journal of Experimental Medicine 96 (1): 83–97. doi: 10.1084/jem.96.1.83
LeBlanc S., Arabzadeh A., Benlolo S., Breton V., Turbide C., Beauchemin N., and Nouvion A-L. 2011. “CEACAM1 Deficiency Delays Important Wound Healing Processes.” Wound Repair and Regeneration: Official Publication of the Wound Healing Society [and] the European Tissue Repair Society 19 (6): 745–52. doi: 10.1111/j.1524-475X.2011.00742.x
van Sorge N.M., Bonsor D.A., Deng L., Lindahl E., Schmitt V., Lyndin M., Schmidt A., Nilsson O.R., Brizuela J., Boero E., Sundberg E.J., van Strijp J.A.G., Doran K.S., Singer B.B., Lindahl G. and McCarthy A.J. 2021. “Bacterial Protein Domains with a Novel Ig-like Fold Target Human CEACAM Receptors.” The EMBO Journal 40 (7): e106103. doi: 10.15252/embj.2020106103
Stålhammar-Carlemalm M., Areschoug T., Larsson C., and Lindahl G. 1999. “The R28 Protein of Streptococcus Pyogenes Is Related to Several Group B Streptococcal Surface Proteins, Confers Protective Immunity and Promotes Binding to Human Epithelial Cells.” Molecular Microbiology 33 (1): 208–19. doi: 10.1046/j.1365-2958.1999.01470.x.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Very nice post! Just to let you know that it seems all the reference links direct to paperpile pages that are not public, so you may want to change that.
Thanks! Updated :)