The multi-omic microverse of SARS-CoV-2 in the lungs of critically ill patients.
Published in Microbiology
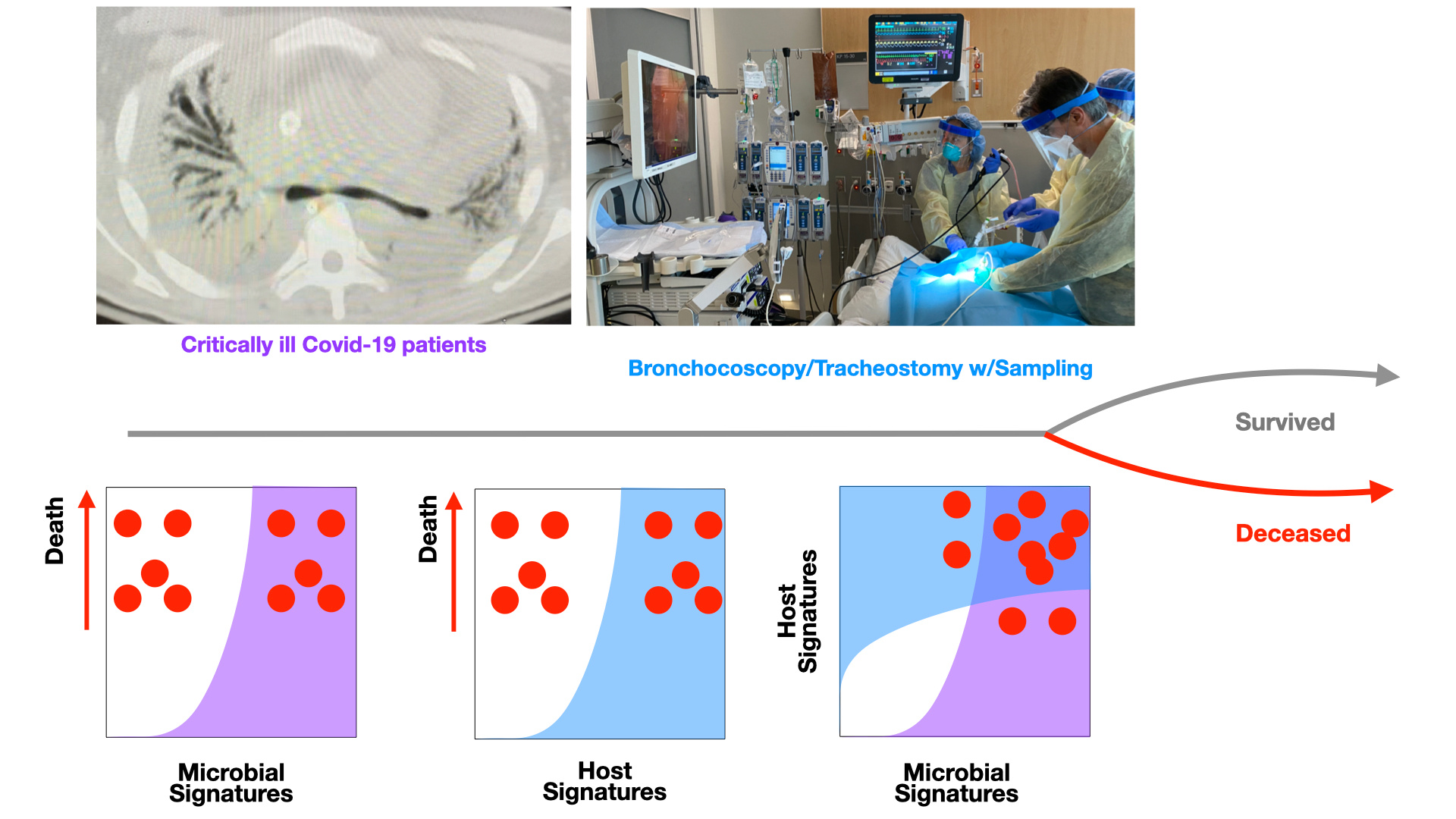

February 2020 everything changed. The world seemed to stop with shut downs and lock downs. However, hospital systems worldwide went into overdrive. This was particularly the case in New York City, where cases of SARS-CoV-2 sky-rocketed during the spring of 2020. At our institution, New York University Langone Health, the volume of critically ill patients exceeded our capacity so much so that we had to create new intensive care beds and garner assistance from across the country to help manage these patients.
I (IS) myself had moved across the Atlantic from Dublin, Ireland to NYC and joined The Segal Lab and the Pulmonary and Critical Care Department of NYU. I split my time between clinical work in the intensive care and research focusing on the lung microbiome. The Segal Lab being pioneers in the exploration of the lower airways microenvironment and its relationship to different respiratory diseases such as lung cancer, bronchiectasis and chronic obstructive pulmonary disease (COPD). Almost naturally we developed a close collaboration with The Ghedin Lab (MC) at the National Institute of Allergy and Infectious Diseases, a genomics group that specializes in the respiratory microbiome in the study of influenza virus infections and, since the outbreak, has been applying their expertise to tackle SARS-CoV-2.
SARS-CoV-2 presented a new challenge to physicians across the world. A novel virus, fiercely spreading globally, infecting and causing the death of millions. Although we had seen other outbreaks—such as the H1N1 2009 pandemic, and the SARS-CoV-1 and MERS epidemics in 2001 and 2012, respectively—this was new territory for all of us. Understanding this virus and its impact on the human host was always going to be crucial to help guide therapies. Since 2012 the use of next generation sequencing (NGS) has become more and more sophisticated. With newer technical instruments and newer bioinformatic platforms the microbial universe was now truly unlocked.
With our prior experience in multi-omic data, The Segal and Ghedin labs set out to study this virus and its effect on the lower airways using next generation sequencing (NGS) with metagenome and metatranscriptome analyses.
Clinical Challenges
Of course, this was not a small feat. Trying to perform potentially clinically important research in the middle of a pandemic when resources are stretched to their limit was challenging, to say the least. Furthermore, collecting lower airways samples, via bronchoscopy (a potentially aerosolizing event) in patients with a highly infectious virus provided extra challenges. However, through this pandemic our critical care team, led by Dr. Luis Angel, developed a new technique for performing percutaneous tracheostomies to cut down aerosolization risk1. This provided us with an ideal window for collecting lower airway samples. In 142 critically ill patients with SARS-CoV-2 infection we were able to collect, process and analyze lower airways samples via bronchoscopy.

Sample Processing Challenges:
As hospitals in New York were being flooded with patients all research efforts across academic institutions ceased except for those directed to this disease. At that crucial time, we had to decide how to adapt to the current situation, who could guide the processing of such infectious samples, what level of containment we would need, how would we do the measurements we needed using equipment not set up for this level of containment? As what probably happened in many academic institutions, the same “all hands on deck” sentiment dominating the clinical wards occurred in the NYU and NIH research community, bringing a broad range of laboratories together to offer BSL3 processing and containment, and multiple different approaches for different measurements that were highly complementary, as shown in this paper.
Data Analysis Challenges:
One important challenge on the analysis front was largely due to the study design: sampling was done as part of the treatment of care of patients who were mechanically ventilated; these patients would go on to meet different fates in their outcomes. Thus, sample access was “opportunistic”. Our challenge was to derive significance from the different signals arising across three separate sets of sequence data from these lower airway samples (host transcriptome, metagenome, and metatranscriptome) and find indicators in each dataset linked to poor clinical outcomes. We needed an analytical design that would identify: (1) key signatures in each of the individual datasets and (2) key interactions across multiple datasets that are linked to poor clinical outcome.
From a technical standpoint, the analysis was complex in that each of the different ‘omics datasets needed to be independently run through different upstream pipelines and downstream analyses while later being integrated for multi-omics analyses. The first portion of our analysis interrogated each dataset independently to find factors linked to poor clinical outcomes. For the host transcriptome we conduced differential expression and pathway analyses to identify biological functions enriched in the lower airways. One of the complexities was making sense of all the microbial, viral, and fungal sequence matches we obtained from the analysis of the metagenomes and metatranscriptomes: Because of likely spurious hits and potential contamination, we experimented with four different methods to identify and mark probable contaminant taxa. Additionally, fine-tuning our data to account for nearest neighbors and reference database biases made distilling this massive amount of information down to a representative picture of the true microbial composition very challenging. We complemented this analysis with functional and resistome profiles to find key signatures in the microbiome of individuals with poor clinical outcomes. These different downstream analyses were all integrated using a multiscale co-expression network analysis, in which data from all three ‘omics datasets were combined to identify larger co-expression modules indicative of microbe-microbe and microbe-host interactions linked to clinical outcomes.
What’s unique
Multiple investigations have compared patients infected with SARS-CoV-2 that have different degrees of severity. Here, we had the opportunity to compare patients with similar disease severity at the time of sampling and identify microbial and host signatures predictive of poor outcome. Further, most investigations have focused on non-invasive samples when what we really want to understand is what’s happening in the primary site of the disease: the lungs. That’s where the power of lower airway sampling with bronchoscopy comes allowing us to identify signatures predictive of mortality that otherwise could not have been found with less invasive samples.

So what did we find:
Armed with these vast datasets we set up twice weekly meetings to digest and discuss. In our meetings, a clear signal appeared quite early; although the clinical spectrum of COVID19 related respiratory failure is extremely heterogenous, the abundance of SARS-CoV-2, by next generation sequencing, had a strong association with poor outcome. We found this signal to also be true with reverse transcription PCR (a direct quantitative method) for the SARS-COV-2 N gene, suggesting that antivirals might still have a role in the treatment of critically ill COVID-19 patients.
Interestingly, we also found that higher abundance of Mycoplasma salivarium (again by NGS), a known oral commensal, was associated with poor outcome. Similar to data previously published by our group, the presence of oral commensals in the lower airways is associated with an inflammatory phenotype. However, importantly, and contradictory to other studies, there was no evidence that co-infection was associated with poor outcome in these patients, that otherwise are frequently treated with broad spectrum antibiotics. Using detailed clinical laboratory culture data available for 589 subjects hospitalized with respiratory failure due to COVID-19, we showed that higher rates of respiratory infection with other organisms, especially early in their hospitalization, did not occur among subjects with poor clinical outcome.
It was also important for us to understand what this viral infection was doing to our patients. In evaluating the host immune response, we measured levels of anti-spike and anti-RBD antibodies in the lower airways. Lower levels of these markers were associated with poor outcome. We also used the NGS data to examine the host transcriptome across the different clinical outcomes. The sirtuin pathway, associated with increased host inflammatory response to viral infection2 and the ferroptosis pathway, a form of non-apoptotic regulated cell death associated with direct lung injury3 were both upregulated in most critically ill COVID-19 patients. Down-regulation of inositol related pathways was also associated with poor outcome. Collectively, these data suggest that an imbalance rather than an elevated inflammatory state in the lung is an important marker that predicts poor outcomes in critically ill COVID-19 patients.
We then wanted to know how well these datasets predict mortality. In a collaboration with the Hulin Lab at NYU, we used each of our ‘omic datasets to model against clinical outcomes. Interestingly, the combination of microbial data (by metatranscriptome) and host transcriptomic data was most predictive of poor outcome, a model that was highly dependent on the relative abundance of SARS-CoV-2.
Where to next:
While this first study primarily focused on samples collected at one specific time-point across a cohort of patients, the next phase of our study is to follow the same individuals with longitudinal sampling. We will also expand our analyses by mapping out the ecological interactions (phages/bacteria, SARS-CoV-2/human, viruses/fungi) that occur in the lower and upper airways. Importantly, this longitudinal sampling will allow us to start dissecting the effects individual therapeutic interventions have on the lower airway microbial and host environment.
References:
- Angel, L. F. et al. Percutaneous Dilational Tracheostomy for Coronavirus Disease 2019 Patients Requiring Mechanical Ventilation. Crit Care Med 49, 1058-1067, doi:10.1097/CCM.0000000000004969 (2021).
- Budayeva, H. G., Rowland, E. A. & Cristea, I. M. Intricate Roles of Mammalian Sirtuins in Defense against Viral Pathogens. J Virol 90, 5-8, doi:10.1128/JVI.03220-14 (2016).
- Qiang, Z. et al. Nrf2 and STAT3 Alleviates Ferroptosis-Mediated IIR-ALI by Regulating SLC7A11. Oxid Med Cell Longev 2020, 5146982, doi:10.1155/2020/5146982 (2020).
Follow the Topic
-
Nature Microbiology
An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in