The S-component fold: a link between bacterial transporters and receptors
Published in Chemistry, Microbiology, and Protocols & Methods
Structure determination of membrane proteins has been a big challenge for decades due to numerous experimental bottlenecks1. Thus, mechanistic understanding of essential processes, such as sensing molecules present in the environment and the transport of metabolites, was slow to emerge. In addition, evolutionary relations between membrane proteins, which often become evident only by comparing of three dimensional structures, have remained elusive because of the limited amount of experimentally determined structures.
In 2021, it became clear that artificial intelligence (AI) could be a game changer in tackling this challenge, as the neural network AlphaFold demonstrated to generate high-accuracy structure predictions of thousands of proteins bypassing the wet lab2. Since then, AI-predicted structures have become plentiful, while other new AI-based tools (e.g., Foldseek, ColabFold, CLEAN, etc.) have laid the foundation for exploring the known protein universe in a completely unprecedented way3.
In the summer or 2023, prompted by the recent release of Foldseek4, we decided to test the capabilities of this novel AI-based tool using the structures of substrate-binding proteins, known as S-components, as entries. S-components are membrane proteins widespread among Gram-positive bacteria5, that bind vitamins and other essential nutrients with high affinity, enabling substrate intake upon the association with a tripartite ATP-hydrolyzing motor, the energy-coupling factor (ECF) module6. The S-component family is poorly conserved in terms of amino acid sequence, but has a characteristic fold of six alpha-helices. Since their discovery in the late 1970s, S-components have been considered as a group of its own, not related to any other protein family. Therefore, searching via Foldseek for structural homologues in the enormous database of structures predicted by AlphaFold, we expected only other S-components among the results. However, a large portion of the hits included 5TMR-sensor histidine kinases (5TMR-SHKs), regardless of the specific S-component used as search query. Using experimentally determined or AI-predicted structures of S-components as queries yielded the same outcome of the resulting Foldseek hits, strengthening the apparent connection with the membrane domain of SHKs.
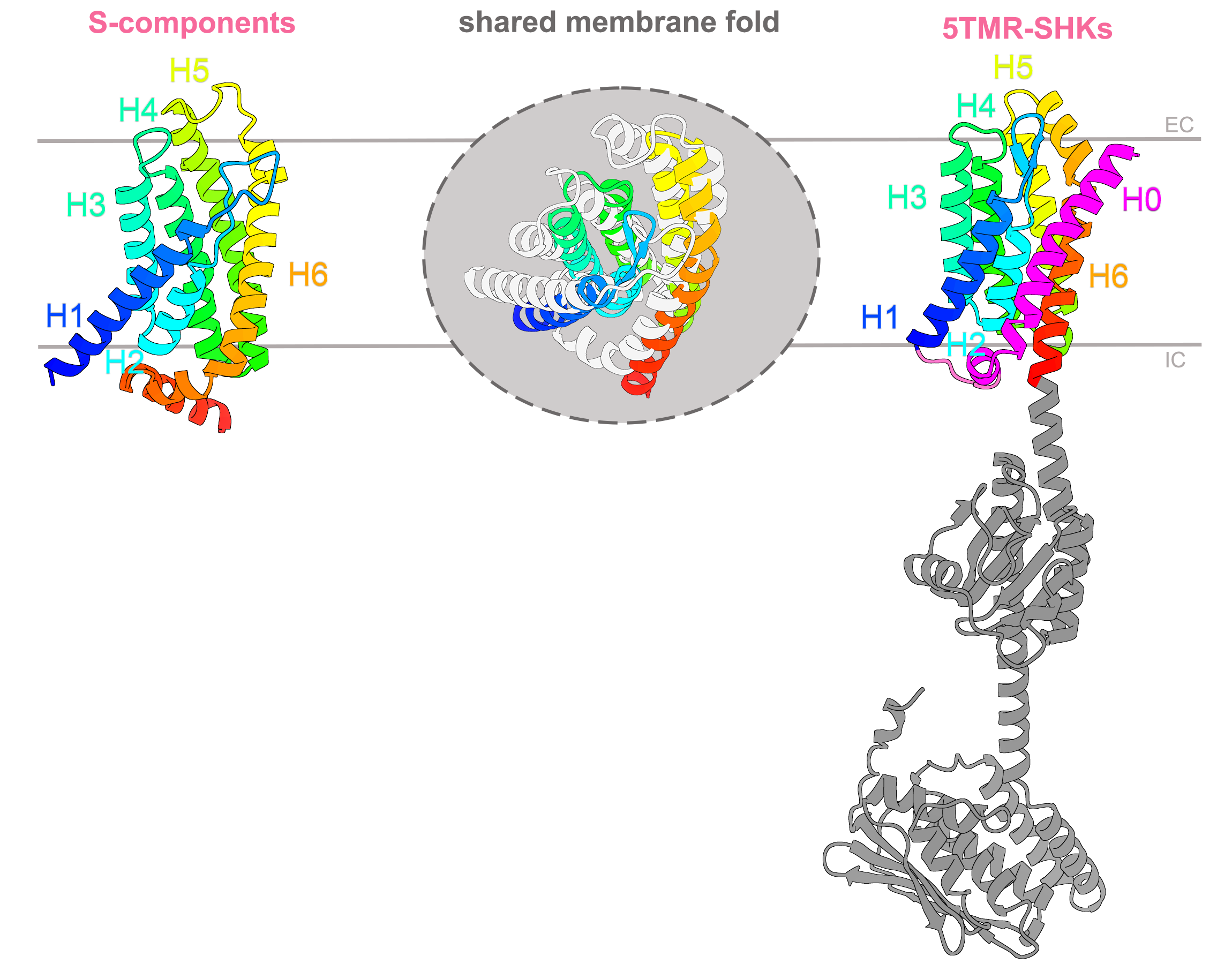
Figure 1 The shared fold between S-components and 5TMR-sensor histidine kinases. Structural similarity search via Foldseek highlighted that substrate-binding proteins involved in membrane transport and the membrane domain of 5TMR-receptors with the function of signal sensing, share the same fold of six membrane-spanning α-helices (H1-H6, in rainbow). The 5TMR-domain has an additional helix H0 at the N-terminus (in magenta), and is linked at the C-terminus to soluble domains needed for signal transduction. The superimposition of the two domains is shown at the center of the figure, with the S-component fold shown in light gray, while helix 0 of 5TMR-fold is omitted. IC and EC indicate the intracellular and extracellular boundaries of the cell membrane. The AlfaFold structures used in this illustration correspond to RibU (identifier AF-Q8Y5W0-F1) and YpdA (identifier AF-P0AA93-F1 ).
This results excitingly revealed that the same fold was shared between proteins with two distinct functions, in nutrient transport and signal sensing (Figure 1). In Eukarya, these two processes are in very rare cases conducted by structurally and evolutionary related proteins, the so-called “transceptors”7, in which receptors have the fold of a membrane transporter. However, transceptors were completely unknown in prokaryotic organisms. At this point, in the attempt to discover what else could link S-components and 5TMR-SHKs, we opted for the integration of bioinformatic approaches with the new AI-tools to expand the insight into the shared membrane fold.
A look into the taxonomic distribution of 5TMR-SHKs and S-components pointed out the widespread distribution of the shared fold among the bacterial kingdom. Surprisingly, more than 50% of the microbes equipped with 5TMR-SHKs are Gram negative, greatly deviating from the abundance of S-components associated with ECF transporters in Gram positive bacteria. The different specialization of the same membrane fold into diverse functions and taxonomic occurrences led us to ask ourselves about its origin. Can we perhaps trace the evolutionary trajectory that led a six-alpha-helical membrane domain to differentiate in function? Or did this peculiar alpha-helical arrangement evolve independently in separate evolutionary contexts, converging eventually into the same domain able to accommodate distinct membrane processes? Unfortunately, neither conventional approaches based on sequence alignment nor structural phylogenetic tools could be of help to find an answer to this exciting question. We will probably need more accurate structure predictions based on AI software, enabling more sensitive structural phylogenetics analyses, likely corroborated by experimental evidence, for instance from additional experimentally determined structures.
5TMR-SHKs were originally annotated in 20038 as receptors with 5 transmembrane alpha-helices presenting a conserved NXR motif, and a signal sequence at the N-terminus linked to the membrane domain via a cytosolic loop. However, the recent AlphaFold-predicted structure showed 7 spanning helices (Figure 2a), and topologically contradicts the proposed function of signal sequence (the cleavage site should have been extracellular and not protruding towards the cytosolic side). A multialignment of 5TMR-SHK receptors led us to reevaluate the role of the N-terminal helix (H0), uncovering potential leucine zipper-like motifs and a relatively well-conserved salt-bridge, both candidate to enable dimerization at the level of the membrane. The AI-prediction of the homodimeric conformation further supported our hypothesis on the function of H0.
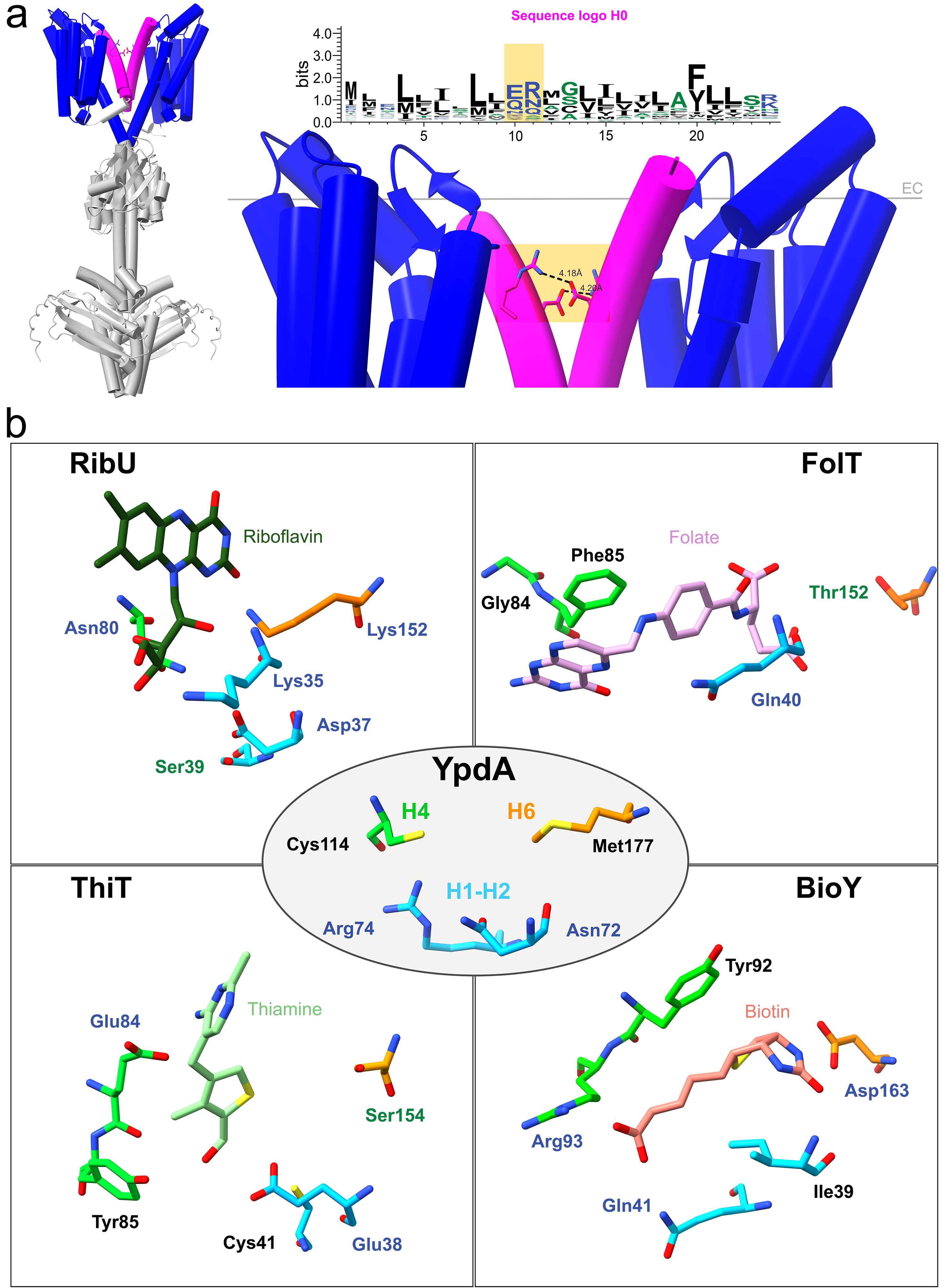
Figure 2 Structural features of 5TMR-sensor histidine kinases. a The role of helix 0 (H0) in receptor dimerization. 5TMR-SHKs have a proposed homodimeric state in vivo, here shown (left of the panel) in the form of the AI-generated prediction of YpdA. While soluble domains (in light gray) enable dimerization of the receptor in correspondence of the regions involved in signal transduction, interactions must also occur at the membrane interphase. The 5TMR-domain (in blue) is linked to an additional helix H0 at the N-terminus (in magenta), which we hypothesized being required for dimerization at the level of the membrane. Besides regularly spaced leucine residues that can establish hydrophobic interactions, H0 has two conserved polar amino acids candidate for the formation of an intramembranous salt bridge (shaded in orange). b Comparison between conserved amino acids in 5TMR-SHKs and amino acids involved in substrate binding for different S-components. The AlphaFold-structure of YpdA from E. coli (PDB entry: AF-P0AA93-F1) was superimposed with experimentally determined structures of S-components RibU, FolT, ThiT and BioY (PDB entries: 5KBW, 4POP, 5D0Y, 4DVE) in complex with their substrates. Asn72, Arg74, Cys114 and Met177 of the receptor YpdA resemble the tridimensional organization of particular amino acid residues needed for substrate binding in different S-components, regardless of their substrate specificity. These amino acids are located in the extracellular loop between helices 1 and 2 (in cyan), helix 4 (green) and helix 6 (in orange), and retain the same orientation towards the substrate pocket, albeit having distinct roles in substrate binding.
We further found highly conserved hot spots in multiple regions facing the extracellular boundary, which structurally align with the substrate-binding sites in experimentally structures of S-components (Figure 2b). This finding is very intriguing because it suggests that the location of the binding site is conserved between S-components of ECF transporters and the 5TMR-SHKs, yet binding triggers very different downstream responses, i.e. either for transport or for signal sensing.
To conclude, we have found a connection between transporters and receptors, two classes of proteins that are generally considered unrelated, revealing a manifestation of a new “transceptor” class in prokaryotes: membrane proteins that have crossed the border between transporters and receptors. Our work showcases how AI-based structure prediction methods lead to new biological discoveries. Our workflow is directly accessible to scientists not specialized in structural biology, and is likely to lead to a plethora of further findings.
References
1. Bill, R. M. et al. Overcoming barriers to membrane protein structure determination. Nat. Biotechnol. 29, 335–340 (2011).
2. Jumper, John, et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
3. Nestl, B. M., Nebel, B. A., Resch, V., Schürmann, M. & Tischler, D. The Development and Opportunities of Predictive Biotechnology. ChemBioChem 202300863, (2024).
4. van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2023).
5. Rempel, S., Stanek, W. K. & Slotboom, D. J. ECF-Type ATP-binding cassette transporters. Annu. Rev. Biochem. 88, 551–576 (2019).
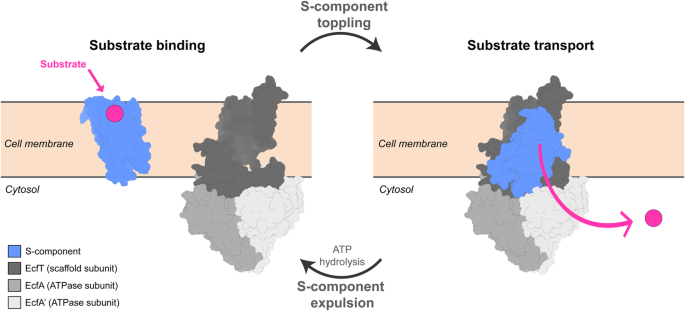
6. Thangaratnarajah, C. et al. Expulsion mechanism of the substrate- translocating subunit in ECF transporters. Nat. Commun. 14, 4484 (2023).
7. Steyfkens, F., Zhang, Z., Van Zeebroeck, G. & Thevelein, J. M. Multiple transceptors for macro- and micro-nutrients control diverse cellular properties through the PKA pathway in yeast: A paradigm for the rapidly expanding world of eukaryotic nutrient transceptors up to those in human cells. Front. Pharmacol. 9, 1–22 (2018).
8. Anantharaman, V. & Aravind, L. Application of comparative genomics in the identification and analysis of novel families of membrane-associated receptors in bacteria. BMC Genomics 4, 1–20 (2003).
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in