The split personality of a molecule under tension
Published in Chemistry

Synthetic polymers are used in almost every aspects of modern life. For this reason, they also present an important environmental challenge. Controlling their mechanical properties at the molecular level could enhance their lifespan and/or improve their recyclability. This goal can be achieved by implementing mechanoresponsive molecules, called mechanophores, that react upon application of a force to generate a useful chemical entity. For example, a force-activated catalyst could be used to repair a damaged material in situ, or to facilitate its degradation at the end of its life. Only a handful of mechanocatalysts have been reported but they’re all metal-based. We wanted to introduce a potentially cheaper and environmentally-friendly metal-free alternative. We settled on a force-activated N-heterocyclic carbene (NHC) precursor (NHCs are very effective organocatalysts) that is inspired by a heat-activated version initially proposed by Hendrick and Waymouth. Our aim was to create a new mechanocatalyst but we ended up uncovering very unusual mechanochemical processes!
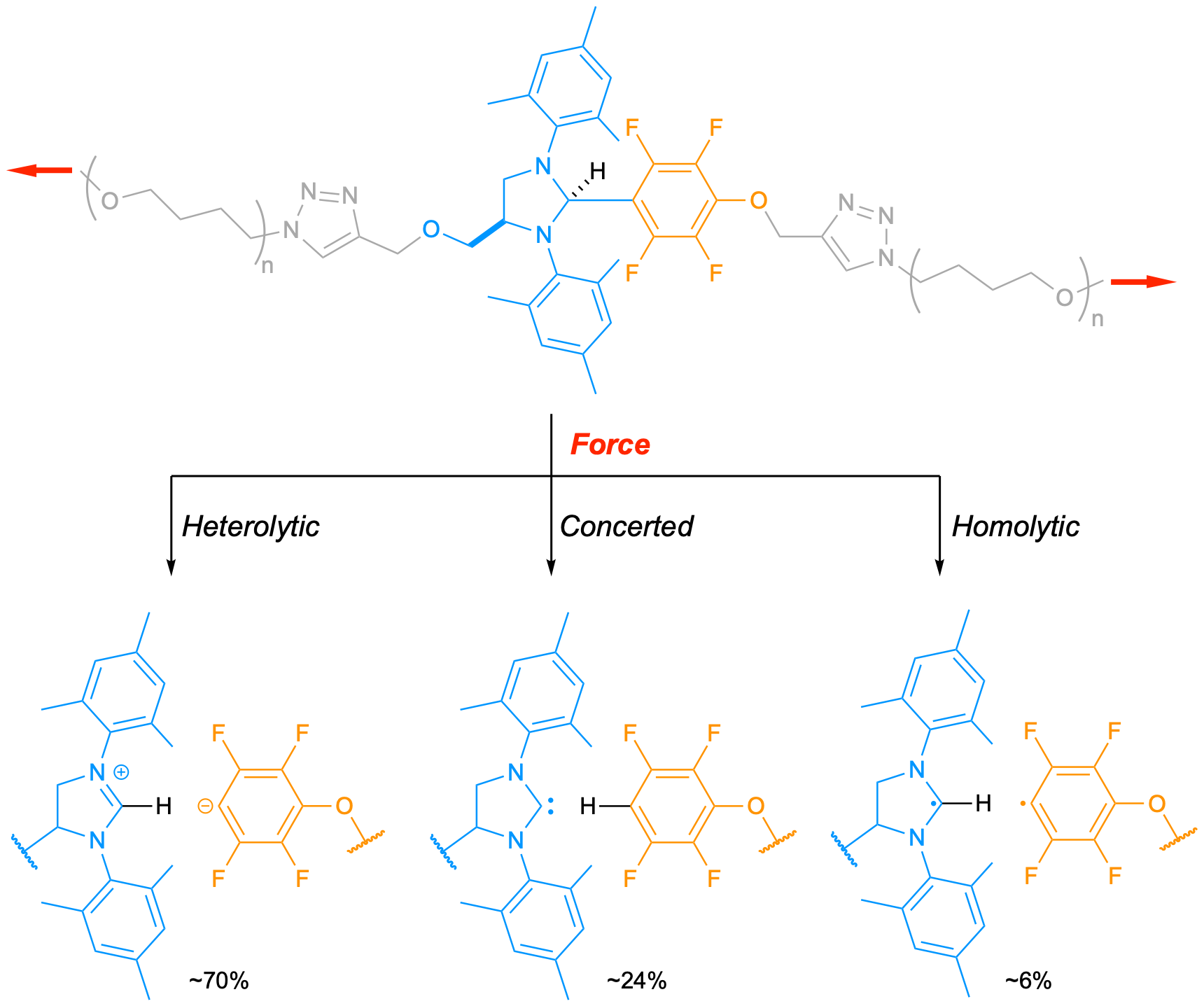
Robert Nixon (first author) was provided with this project for his PhD. He quickly managed to obtain the mechanophore and incorporate it into a poly(methyl acrylate) (PMA) backbone. Analysis of the post-sonication mixture of the polymer-mechanophore adduct (ultrasound-induced cavitation generates elongational flows that stretch polymers in solution), suggested a selective cleavage of the desired C-C bond in the mechanophore and we assumed that the desired catalytically active carbene was formed in the process. Unfortunately, whatever reaction we tried, no catalysis took place. As we feared that the carbene could react with the pendant ester groups of the PMA, we decided to use polytetrahydrofuran (PTHF) instead. PTHF has the advantage to be mechanically robust, and inert to carbenes. Despite this change, we still couldn’t get any catalysis to work. One day, Rob arrived in my office to chat about the project and, at the end of the discussion, mentioned a particularly well resolved 1H NMR spectrum he had obtained after his latest sonication. ‘Do you want to see it?’, he asked. ‘Sure’, I replied (I always enjoy looking at nice NMR spectra). We talked a bit about how clean the spectrum was until we spotted the critical triplet of triplets, unresolved until then, which is diagnostic of the lonely proton in the tetrafluorobenzene derivative. This observation finally confirmed that the desired C-C bond was cleaved during the activation! It also meant that the bond cleavage didn’t proceed via the desired concerted pathway and/or that the carbene was quickly quenched after formation.
After ruling out any premature quenching of the carbene, we focused our attention on the dissociation mechanism(s) itself. Luckily, the presence of fluorine atoms on the aromatic leaving group allowed us to use 19F NMR, which greatly facilitated our investigation. Indeed, these F atoms display a slightly lower shift when they are adjacent to a D instead of H. We performed the mechanical activation in presence of various sources of D (in the mechanophore, D2O, or THF-d8) and found that the scissile C-C bond was cleaving following heterolytic, homolytic, and concerted pathways… at the same time! It was the first time that such a triple dissociation was observed in mechanochemistry. We then showed that we could control the outcome of the dissociation by altering the polarisation of the scissile bond (fewer F atoms on the aromatic favours the concerted pathway). The last piece of the puzzle was to understand how these three dissociations pathways could coexist. We performed a series of calculations to probe the effect of mechanical force on the shape of the potential energy surface (PES) and found the product distribution is the result of a post-transition state bifurcation. Namely, the downward slope of the PES divides into two valleys upon application of a force, one leading to the heterolytic product and the other to the concerted products. This was a surprising result for us because post-transition state bifurcations are not very common in organic chemistry, let alone in polymer mechanochemistry.
As is often the case in science, we learned much more than anticipated at the start of the project. We haven’t been able to catalyse a reaction with our carbene (yet!), but we’ve uncovered some unprecedented mechanochemical reactivity and shown how to control it to alter the outcome of the dissociation. The ability to produce such a variety of reactive species on demand should open new avenues in the design of mechanoresponsive materials.
Group website: https://www.deboresearchgroup.com
Follow us: @GuillaumeDebo
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in