TISSUE: Predicting missing spatial gene expression with confidence
Published in Computational Sciences and Genetics & Genomics
Spatial transcriptomics is a new and exciting technology, allowing scientists to measure gene expression, sometimes at single-cell resolution, while preserving the spatial context of the biological sample. These approaches have provided new insights into spatial biology—patterns of expression during development and aging, cell-cell interactions, and cell neighborhoods underlying different phenotypes.
What motivated us to build TISSUE
Before we even considered developing TISSUE, we were interested in collecting spatial transcriptomics data on the adult mouse subventricular zone, a region of the brain that is resident to neural stem cells (NSCs). At that time, NSCs and their lineage had not been profiled using spatial transcriptomics so we selected our own marker genes for these cell types from existing single-cell RNAseq atlases and genes coding for known NSC markers. After performing a small-scale spatial transcriptomics experiment, we were dismayed to find that these markers were insufficient for identifying the cell types of the NSC lineage at the resolution that we desired.
Between a rock and a hard place
We seemingly had two options to move forward with the project, neither of which was particularly attractive at the time. The first option would be to redo the spatial transcriptomics experiment with a larger and redesigned gene panel, which would hopefully capture the NSC lineage cell types that we were interested in. This would be resource-intensive in both time and money. The second and more cost-efficient option would be to leverage computational methods for predicting spatial gene expression to augment the experimentally measured marker genes. We implemented several existing algorithms but were unable to confidently identify the cell types of interest even after imputing the expression of many additional marker genes.
A large part of this uncertainty revolved around the predicted expression of the marker genes. How accurate were the predictions? Could we trust them? This issue was further compounded by the high variability of prediction performance across different genes or cells. This meant that a method could make poor predictions for a gene and cell type even if it had great performance on other genes and cells. If only there was a way to know which predictions to trust!
The TISSUE framework
Seeing that there was no universal method for estimating uncertainty for spatial gene expression predictions, we set out to build our own method. We borrowed ideas from conformal inference, an approach in machine learning for calculating well-calibrated measures of uncertainty for model predictions, to generate a framework for estimating uncertainties for predicted spatial transcriptomics. We named this framework TISSUE (Transcript Imputation with Spatial Single-cell Uncertainty Estimation).

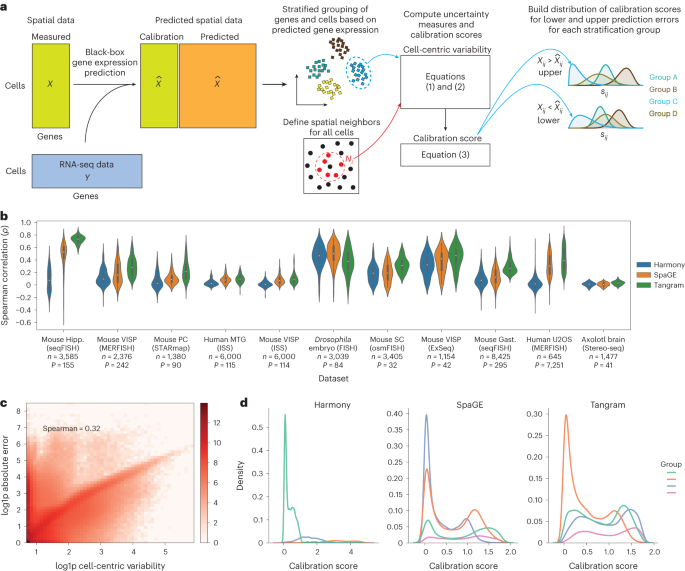
The first objective of TISSUE is to calculate a prediction interval for any given gene and cell in a spatial transcriptomics dataset. This interval contains the true gene expression at a specified level of probability. To build this interval, TISSUE defines a cell-centric variability measure based on spatial variability in the predicted gene expression and then calibrates this measure to known prediction errors obtained on the spatial transcriptomics gene panel (see Figure 1). An advantage of TISSUE is that it produces these estimates in a context-specific manner (i.e. within a single spatial transcriptomics dataset) and without any need for additional training. TISSUE produces well-calibrated prediction intervals across a wide range of publicly available spatial transcriptomics datasets.
We wanted to extend the usability of TISSUE beyond just estimating uncertainties for predictions and developed several additional methods for downstream analysis. For example, we performed multiple imputations with TISSUE to leverage estimated uncertainties in differential gene expression analysis. This approach resulted in a consistent reduction in the false discovery rate. We also extended TISSUE to cell clustering, model training, and data visualization tasks. For an in-depth summary of TISSUE and its applications, refer to our original manuscript linked at the start of this blog post.
After developing TISSUE, we revisited the problem we encountered in identifying cell types of the NSC lineage in our spatial transcriptomics dataset. Applying TISSUE allowed us to annotate subclusters of cells in the NSC lineage and identify all cell types of interest. To our knowledge, this is the first time that these cell types have been identified using spatial transcriptomics.
We believe that TISSUE can find similar utility in answering other research questions in spatial biology and beyond. TISSUE is available as an easy-to-use Python package.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in