Tracing the history of the second plague pandemic through ancient DNA
Published in Microbiology

Our paper published in Nature Communications can be found here:
https://doi.org/10.1038/s41467-019-12154-0
The second plague pandemic, caused by the bacterium Yersinia pestis, began with the infamous Black Death (1346–1353 AD) and was arguably one of the most devastating pandemics of human history. Despite the ubiquitous traces that it has left on the archaeological and historical records across Europe, still little is known about the pandemic’s initiation and progression between the 14th and 18th centuries.
How did the bacterium enter Europe? Which route did it take and how did it further evolve during its subsequent spread?
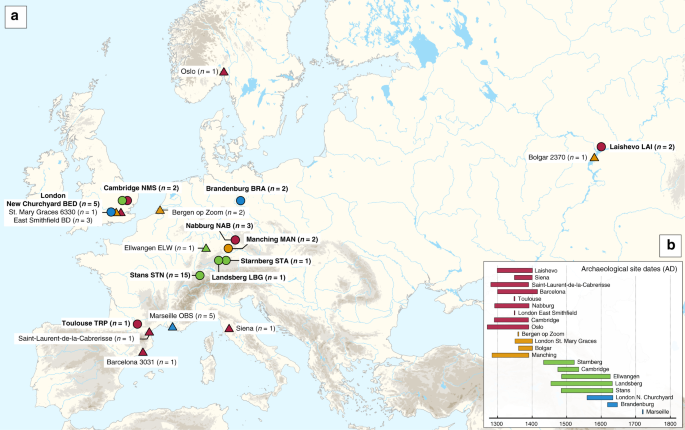
We set out to find clues to these questions by analysing human remains from archaeological sites spanning the 14th to 17th centuries with the aim of identifying traces of Y. pestis DNA. Few excavated sites are known plague cemeteries, so, often, mass or multiple burials are only indicators of unspecific epidemic events. As a result, we encountered collections where none of the tested individuals were positive for Y. pestis, either because of a different epidemic cause or due to poor DNA preservation −a pervasive challenge in ancient pathogen research. Ultimately, our combined efforts resulted in a dataset that consisted of 10 medieval and post-medieval archaeological sites in England, France, Germany, Russia, and Switzerland, from which we could reconstruct 34 ancient Yersinia pestis genomes.

Locations and chronology of new (circles) and previously published (triangles) Y. pestis genomes from the second plague pandemic. Appears as Figure 1 of the paper.
So what did we find?
Our first conclusion was formed through the analysis of a 14th-century archaeological site in the Volga region of Russia. Here, we could retrieve DNA from two plague victims, and isolated Y. pestis genomes that we identified as the most ancestral variant of the second pandemic so far sequenced. This result shows that Y. pestis entered Europe through the east; a notion that has for long been portrayed in historical records.
Furthermore, our analysis of new and previously published genomes from the Black Death period in the regions of modern-day England, France, Germany, Italy and Norway showed that they are all identical. This suggests that the initial wave of the pandemic was caused by a single Y. pestis strain that dispersed rapidly across the continent without accumulating any genetic diversity.

Mass grave of the Black Death period, identified in the “16 rue des Trente Six Ponts” archaeological site in Toulouse, France. ©Archeodunum SAS, Michaël Gourvennec.
Furthermore, based on historical records and previously published ancient DNA research, we know that, after the Black Death, plague continued to cause recurrent outbreaks in Europe until the 18th century. In order to further investigate this time period, we sampled multiple post-Black Death epidemic sites, with a particular emphasis on western and central Europe. Here, we could retrieve a diverse pool of strains that were responsible for epidemics between the late-14th and 17th centuries. These strains were found to be descendants of the initial Black Death strain, hence, suggesting the local persistence and diversification of Y. pestis in Europe after 1353.

Plague burial of three juvenile individuals from Starnberg, Germany, dating to the 15th-16th centuries. ©Bavarian State Department of Monuments and Sites.
Finally, a yet mysterious riddle in our story arose during the analysis of genomes from the late stages of the pandemic. Specifically, we identified a large deletion, including virulence-associated genes, among 17th and 18th century isolates. Given that this deletion was found in strains associated with epidemic events in London and Marseille, we believe that it is unlikely to have affected the bacterium’s pathogenicity in humans.
So, which could be its functional consequences?
Given that the genomes that show this deletion are part of an extinct lineage −consistent with plague’s disappearance from Europe during the 18th-19th centuries− our first idea was to check whether an identical deletion was present among other ancient or modern Y. pestis genomes in our dataset. To our surprise, we could only identify it among genomes associate with the first plague pandemic (6th- to 8th- centuries AD), which were also found to be part of an extinct lineage. Therefore, there is no surviving Y. pestis that carries such genomic characteristics today. As such, we think that an exciting area for future research would be to determine how these changes affected the bacterium’s ability to infect and be maintained in mammalian and arthropod hosts.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in