Tumor-specific total mRNA expression: a robust and prognostic feature across cancers
Published in Bioengineering & Biotechnology
In the identification of clinically relevant gene signatures across cancers, a common assumption made during RNA sequencing analyses is that tumor cells from different cancer specimens share the same mRNA content. This assumption can result in the biased identification of genes that vary in concentration across patient samples and the omission of genes that change in absolute mRNA abundance. Moreover, total mRNA content is believed to be an important metric on its own and correlated with cellular phenotype.
Measuring total mRNA content in tumor cells is a long-standing topic in cancer. Varying tumor cell total mRNA expression has been linked with stemness and high plasticity at a single-cell level by multiple studies since 20201,2. However, its role in tumor development and progression requires further large-scale studies in clinically relevant samples.
In single-cell RNA sequencing studies, the total mRNA content of tumor cells can be estimated using unique molecular identifiers. However, high costs and sample requirements currently prohibit scalable use of this technology for association studies in large patient cohorts. In addition, single-cell technologies have only recently been implemented to profile clinical tumor samples. Survival outcomes for these patients will still take years to collect, which limits the clinical utility of this sequencing data.
With its cost effectiveness and comprehensive coverage across the genome, bulk RNA sequencing remains the best available method for evaluating tumor transcriptomes in patient samples with clinical outcomes. However, estimation of total transcript levels using bulk RNA sequencing has been limited by several analytical factors including:
- The need to account for technical artifacts introduced by varied library size, which currently involves normalization procedures across samples
- Total transcript counts are confounded with technical artifacts so that normalization procedures adjust for both effects at once, consequently losing the ability to evaluate the global transcriptome feature downstream
- A limited focus on estimating cell proportions by popular methods
Furthermore, bulk RNA sequencing data includes transcript counts from both tumor and non-tumor cells. As such, tumor-specific transcript levels cannot be directly estimated without deconvolution methods to further dissect the cellular populations.
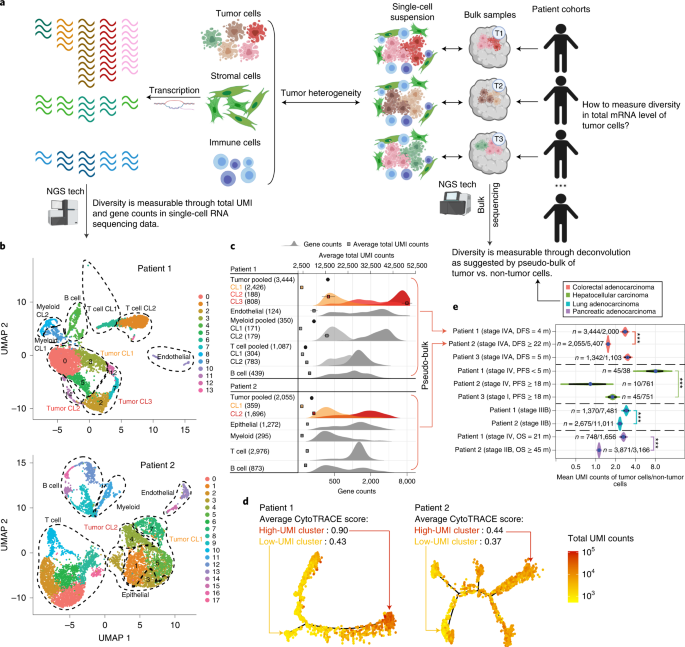
We sought to develop a strategy to overcome these challenges and estimate tumor-specific total mRNA expression in bulk patient samples. Building upon our prior work3,4, we found that using deconvolution methodology to partition tumor and non-tumor cells within the same sample under the same experimental conditions provides a mathematical means to cancel out confounding technical artefacts while maintaining the effect of cell-type-specific total transcript counts.
Our developed metric, termed TmS (tumor-specific total mRNA expression), captures the ratio of total transcript level per haploid genome copy in tumor cells versus surrounding non-tumor cells5. TmS utilizes information obtained from matched bulk DNA and RNA sequencing data. Tumor cell purity and ploidy are obtained from DNA sequencing data and are used to systematically model the expression pattern of bulk RNA sequencing data to quantify the transcript levels of tumor cells.

Figure 1. Analysis workflow to measure tumor-specific total mRNA expression.
In this study, we estimated TmS using large collections of sequencing data from bulk tumor samples where long-term clinical annotations were available, allowing for a comprehensive examination of the associations with clinicopathologic features, molecular phenotypes and clinical outcomes across cancers. Our findings suggest that TmS summarizes tumors’ average transcriptional program. Further analysis across 15 cancer types found that high tumor-specific total mRNA expression appear to generally promote tumor progression, but it may become disadvantageous in certain molecular and/or treatment contexts.
In recent years, histopathologic and molecular features have been identified in many cancer types, which guide risk stratification and treatment selection beyond stage6,7. However, significant variability in clinical outcome remains, and additional refinement in prognostication is needed. We were excited by our findings that TmS has the ability to refine prognostication in all the three large cancer patient cohorts: The Cancer Genome Atlas (TCGA) projects, the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) study and the Tracking Cancer Evolution through therapy (TRACERx) study. Therefore, TmS has the potential to serve as a prognostic biomarker across many cancer types. Additional prospective studies are required to further ascertain how TmS can be used to inform clinical risk stratification and treatment selection across cancers.
In summary, our findings suggest that enhanced attention to global features of the transcriptome will not only contribute to a deeper biological understanding of cancers, but may prove to be a critical step in improving risk stratification, treatment selection and prognostication.
References:
- Marjanovic, N. D. et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell38, 229-246.e13 (2020).
- Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 367, 405–411 (2020).
- Ahn, J. et al. DeMix: Deconvolution for mixed cancer transcriptomes using raw measured data. Bioinformatics29, 1865–1871 (2013).
- Wang, Z. et al. Transcriptome Deconvolution of Heterogeneous Tumor Samples with Immune Infiltration. iScience 9, 451–460 (2018).
- Cao, S. et al. Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression. Nature Biotechnol (2022) . doi: 10.1038/s41587-022-01342-x.
- Wang, X. et al. Predicting gastric cancer outcome from resected lymph node histopathology images using deep learning. Nat. Commun. 2021 121 12, 1–13 (2021).
- Paik, S., Shak, S., Tang, G., Kim, C., Baker, J., Cronin, M., Baehner, F.L., Walker, M.G., Watson, D., Park, T. and Hiller, W. A Multigene Assay to Predict Recurrence of Tamoxifen-Treated, Node-Negative Breast Cancer. N. Engl. J. Med. 351, 2817–2826 (2004).
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in