Understanding the origin of the eukaryotic cell: gene duplications to the rescue
Published in Ecology & Evolution
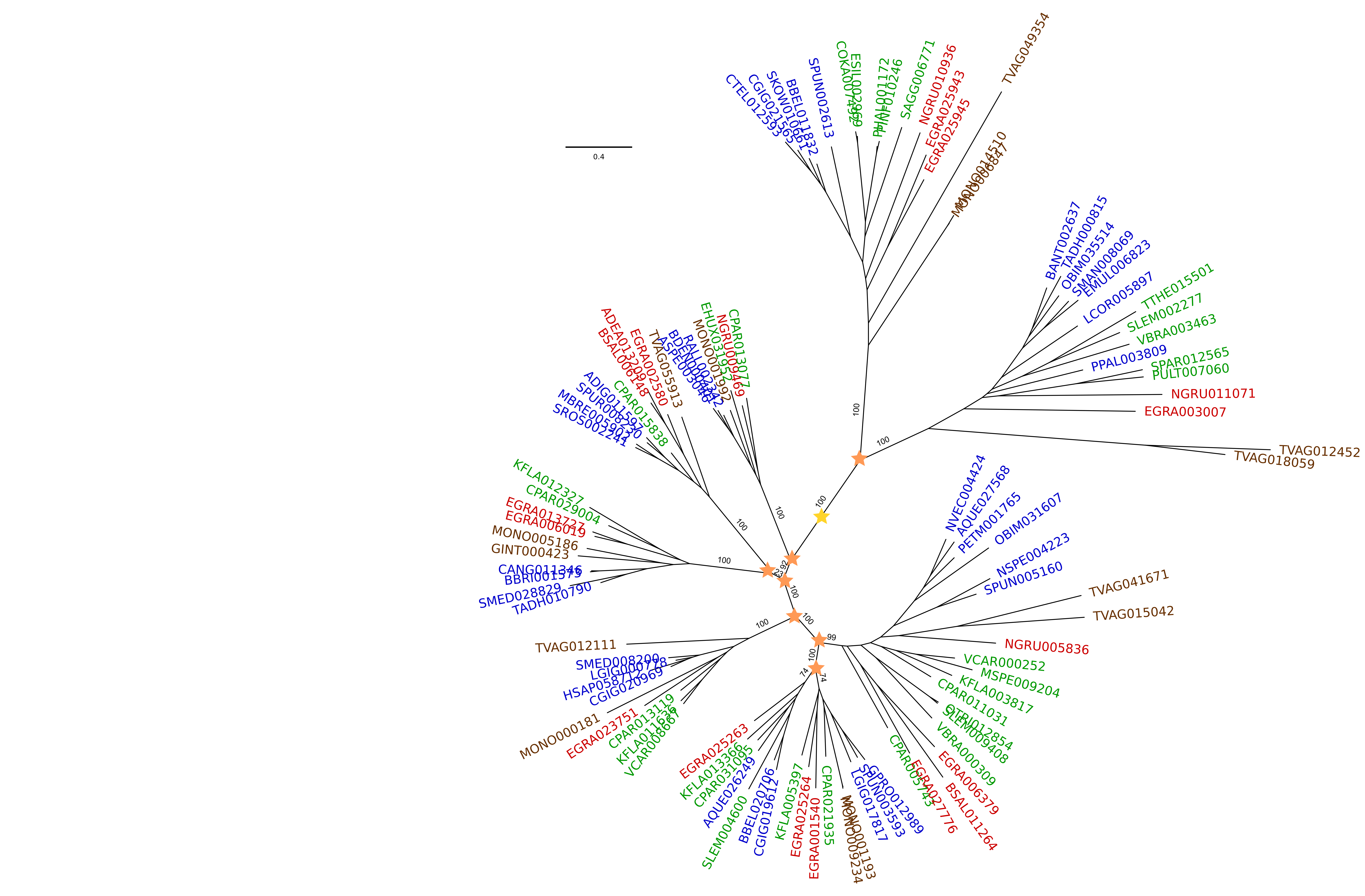

Many papers come by during journal clubs. Most of them do not leave a lasting impression, but from time to time you have a lively discussion about a paper that is the starting point of a new project. In our case it was the article by Pittis & Gabaldón in 2016.
Using branch lengths in gene trees, Pittis & Gabaldón inferred the order in which the primordial eukaryotic genome acquired new genes from different prokaryotic lineages. However, as we discussed during our journal club, another main contributor to eukaryotic complexity were the numerous gene duplications, which were not included in their examination. In fact, after the pioneering work of Makarova et al. in 2005, few large-scale investigations into proto-eukaryotic gene duplications had been published. We wondered: could we use a similar approach to time these duplications and consequently infer a more complete order of events that resulted in eukaryotic cellular complexity?
We contacted Toni Gabaldón, who was willing to collaborate. Together we reasoned that placing duplications onto the eukaryogenesis timeline is difficult. There are no early-branching (proto-)eukaryotic lineages and thus one cannot map duplications either before or after their divergence. However, a branch length-based timing as pioneered by Pittis & Gabaldón might actually work..
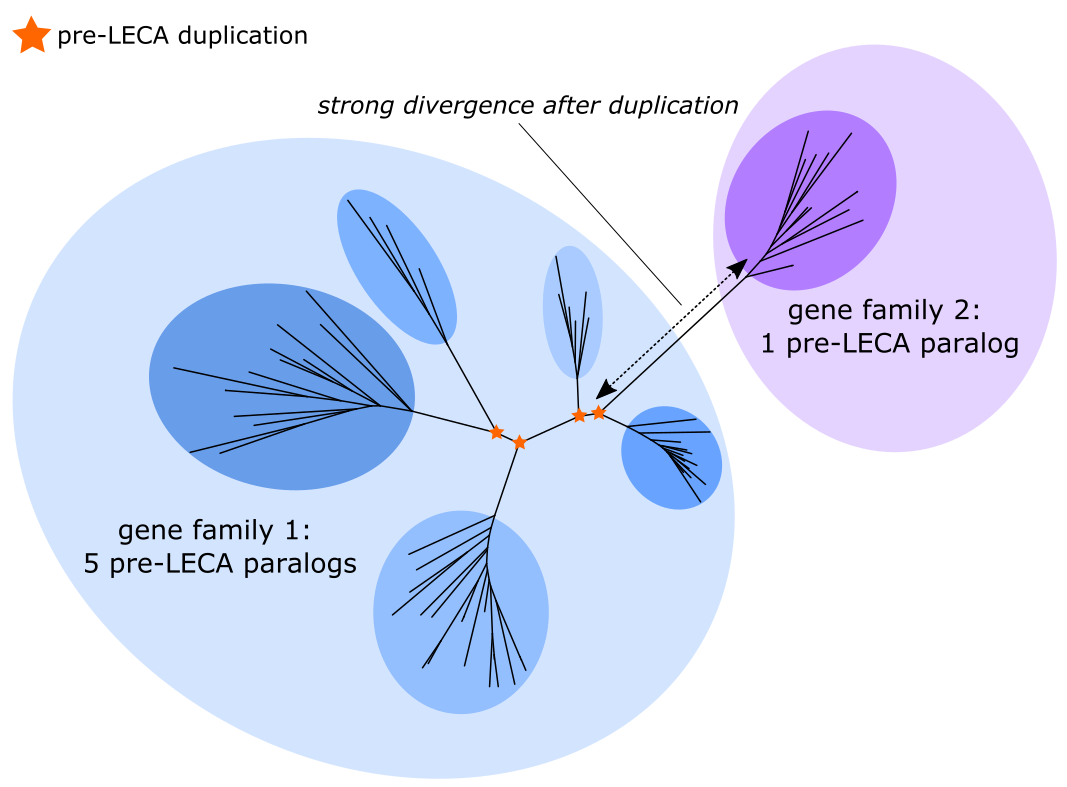
Initial analyses with the original trees of Pittis & Gabaldón yielded very few ancient duplications. This low yield is likely caused by ‘oversplitting’ of highly diverged paralogs into different families and trees (see illustration below). We tried various gene family databases, but, although old, the original Makarova and Koonin database (2005) actually was one of the few that delivered a substantial number of duplications. However, we also knew of various gene families that were not included in their set. We therefore opted for the comprehensive Pfam protein domain database, whose profile HMMs were able to capture distant, strongly diverged homologs.
After we had selected our gene family resource, we were confronted with the inherent difficulties that come along with inferring high-quality gene trees for massively expanded families. Our group previously tackled this problem in an in-depth analysis of the - highly expanded - eukaryotic kinase family by applying ScrollSaw to select slowly evolving sequences. This smart sequence selection strategy increases the number of well-supported deep nodes in phylogenetic trees.

We were ultimately able to deduce many gene duplications during eukaryogenesis. Although these duplications occurred throughout the transition, the data showed clear functional differences. For example, cytoskeletal families duplicated early and signal transduction families relatively late. While the discussion about eukaryogenesis usually focuses on early versus late acquisition of the mitochondrion, our analysis highlights that more intermediate stages can be distinguished. Mitochondrial endosymbiosis was neither the prologue nor the finale, but probably a plot twist in-between.
While we estimated a near doubling of the genome, the number of duplications is very likely an underestimation. For example, multiple ancient duplications that contributed to the emerging kinetochore were not covered in this large-scale approach. The trees constructed in our study can, however, serve as a good starting point to illuminate the evolution of other complexes and processes that originated during eukaryogenesis, such as chromatin modifiers, meiosis, motor proteins and many more.
Follow the Topic
-
Nature Ecology & Evolution
This journal is interested in the full spectrum of ecological and evolutionary biology, encompassing approaches at the molecular, organismal, population, community and ecosystem levels, as well as relevant parts of the social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Understanding species redistributions under global climate change
Publishing Model: Hybrid
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in