Quasicrystals, with their non-repeating, aperiodic structures, have captivated scientists since their discovery in 1982 by Dan Shechtman. Unlike traditional crystals that possess translational symmetry, quasicrystals exhibit long-range orientational order without periodicity, resembling patterns found in Penrose tilings. Despite their mathematical elegance and intriguing physical properties—such as low thermal conductivity, high hardness, and corrosion resistance—quasicrystals remain elusive in terms of practical applications due to challenges in synthesis, stability prediction, and structural characterization.
A promising route to understanding quasicrystals lies in studying their crystalline counterparts, known as approximant crystals (ACs). These materials share local atomic arrangements with quasicrystals but are periodic, making them more amenable to conventional crystallographic analysis. In particular, Mackay-type 1/1 approximants, which contain rhombic triacontahedron (RTH)-based clusters, offer a gateway to exploring the complex intermetallic systems where quasicrystals emerge.
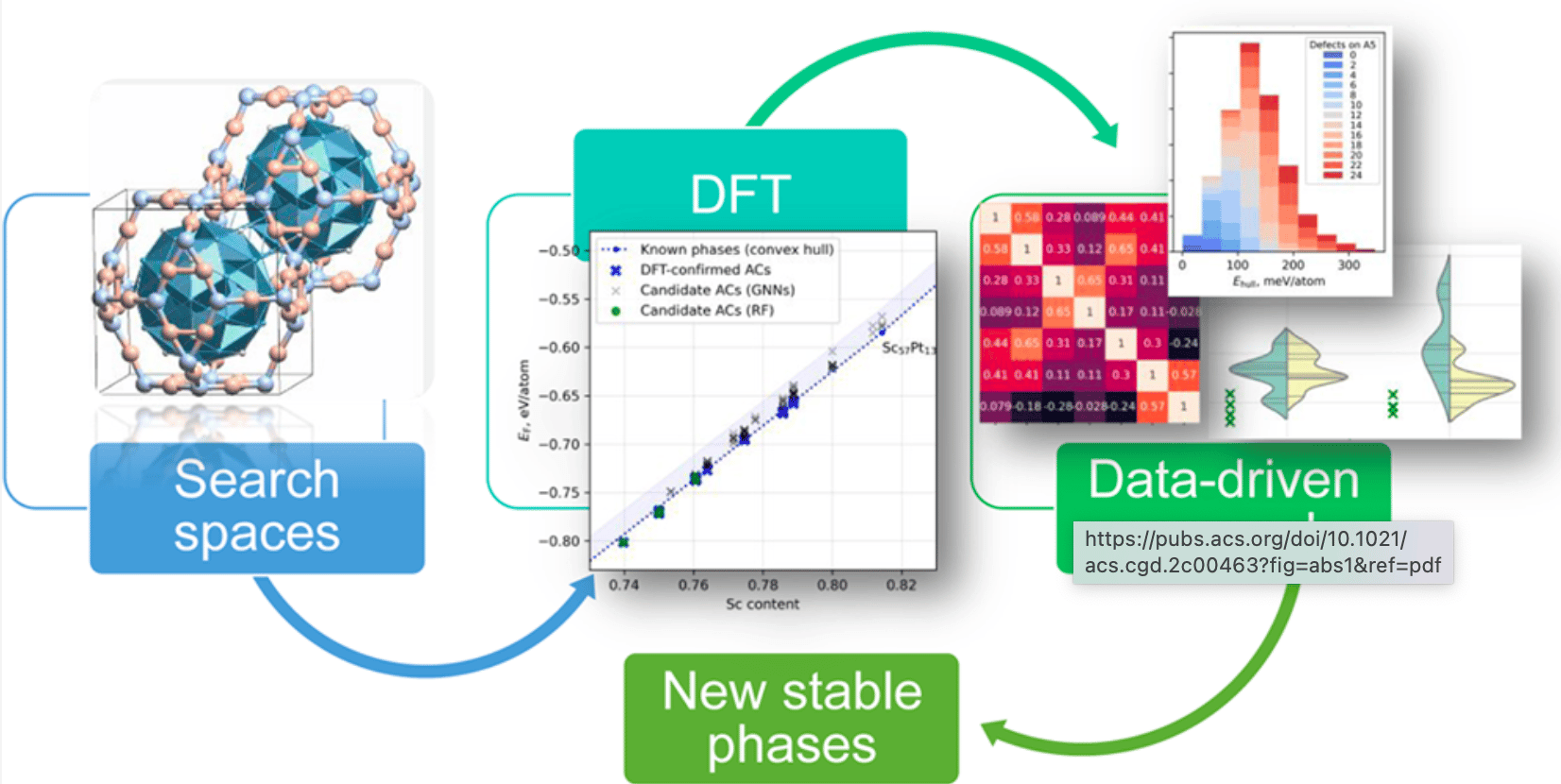
In study published in Crystal Growth & Design , we present a hybrid computational framework combining density functional theory (DFT) and data-driven machine learning techniques to predict new stable 1/1 Mackay-type approximants in Sc–X (X = Rh, Pd, Ir, Pt) systems. This work not only advances our understanding of quasicrystal formation but also paves the way for accelerated materials discovery through intelligent design strategies.
The Challenge: Predicting Complex Intermetallic Structures
Intermetallic compounds—alloys formed between two or more metallic elements—are renowned for their structural complexity and diversity. Many of these materials feature unit cells containing hundreds of atoms, making them difficult targets for conventional structure prediction methods such as evolutionary algorithms or random sampling. Moreover, the presence of disorder, multiple substitutional sites, and competing phases further complicates thermodynamic modeling and stability prediction.
For quasicrystals and their approximants, the challenge is even greater. Due to their intricate cluster-based architectures and often subtle energetic differences between configurations, identifying the most stable structures computationally requires extensive exploration of configuration spaces. Traditional high-throughput screening approaches quickly become computationally prohibitive when applied to large supercells and complex compositions.
Eremin et al. tackle this problem head-on by introducing a novel hybrid methodology that integrates first-principles calculations with advanced machine learning models, significantly reducing the computational burden while maintaining predictive accuracy.
Methodology: A Synergistic Approach Combining DFT and Machine Learning
At the core of the authors’ strategy is the generation of simplified composition/configuration spaces (CCSs), which serve as training datasets for machine learning models. Starting from the experimentally known Sc57Pt13 structure—a prototype of the 1/1 Mackay-type approximant—they introduce controlled defects (vacancies and substitutions) on specific atomic sites within the defect RTH shell. By varying the number and type of defects systematically, they construct simplified CCSs containing up to ~2100 configurations per system.
These simplified datasets are then evaluated using DFT to compute key thermodynamic quantities: relaxed energy, formation energy, and energy above the convex hull (EHull)—the latter being particularly relevant for assessing phase stability relative to competing structures.
With this foundational dataset in place, the team employs two distinct machine learning paradigms:
-
Random Forest (RF) Regression : Using handcrafted descriptors based on defect content and local structural motifs, RF models are trained to predict formation energies and EHull values. These descriptors encode information about the arrangement of defects within the intercluster volume, capturing essential topological features of the Mackay clusters.
-
Graph Neural Networks (GNNs) : Moving beyond manually engineered features, the authors utilize graph-based deep learning models—specifically, SpinConv—to directly learn structural representations from crystal graphs. These GNNs take into account the full connectivity of atoms in the unit cell and are invariant under rotational transformations, making them ideal for modeling complex intermetallics.
By pretraining on the Open Catalyst Project (OCP) dataset and fine-tuning on the DFT-derived energies, the GNNs achieve high recall in identifying low-energy configurations, outperforming RF models in some cases.
Results: Predicting and Validating New Approximants
Applying this hybrid approach across four Sc–X systems (Rh, Pd, Ir, Pt), the authors identify over 20 potentially new stable approximants—most notably in the Sc–Pt and Sc–Pd systems. For example, in Sc–Pt, the union of predictions from RF and GNN models yields 120 candidate structures, of which 23 are confirmed via DFT relaxation to lie within 2 meV/atom of the convex hull—indicating high thermodynamic stability.
Among the predicted structures are both cubic and lower-symmetry variants, demonstrating the method’s ability to capture diverse structural motifs. Notably, one new cubic Sc–Pd approximant was identified, highlighting the potential of the model to uncover previously unknown phases even in well-studied systems.
Importantly, the authors emphasize that different machine learning architectures yield complementary results. While RF models provide robustness and interpretability, GNNs excel at generalization and recall, enabling the discovery of structures missed by simpler approaches.
Discussion: Implications for Materials Discovery and Design
This work exemplifies the power of integrating domain-specific knowledge with modern machine learning tools to accelerate materials discovery. By leveraging the hierarchical organization of intermetallic structures—particularly those involving RTH-based clusters—the authors demonstrate how informed search strategies can dramatically reduce the effective dimensionality of the configuration space.
One of the most compelling aspects of the study is its focus on enhanced configuration spaces , where machine learning models trained on simplified datasets are used to infer the stability of thousands of unexplored configurations. This “search space expansion” paradigm enables researchers to explore regions of chemical space that would otherwise be inaccessible due to computational constraints.
Moreover, the comparative analysis of different target energies (relaxed energy, formation energy, and energy above the convex hull) underscores the importance of selecting appropriate metrics for guiding discovery. As shown in the paper, the performance of GNNs depends critically on the choice of target, suggesting that multi-objective optimization frameworks may be necessary for future efforts.
Finally, the authors highlight the limitations of current descriptor-based models and advocate for the broader adoption of descriptor-free approaches like GNNs, which can automatically extract meaningful features from raw crystal structures without human intervention. This shift toward end-to-end learning aligns with trends in other domains of materials science and promises to unlock new avenues for discovering functional materials with tailored properties.
Conclusion
The study represents a significant step forward in the quest to understand and predict complex intermetallic structures. By fusing DFT-based thermodynamics with cutting-edge machine learning techniques, the authors have demonstrated an efficient and scalable pipeline for identifying stable approximant candidates in Sc-rich systems.
Beyond the immediate scientific impact, this work offers a blueprint for future studies in materials design, especially in areas where high configurational complexity and limited experimental data pose barriers to discovery. As the field moves toward increasingly autonomous workflows—where machines assist in hypothesis generation, experiment planning, and property prediction—hybrid approaches like the one described here will play a central role.
Ultimately, the path to realizing the full potential of quasicrystals and their approximants lies not just in uncovering new structures, but in building intelligent systems that can navigate the vast landscape of possible materials. With continued advances in computation, data science, and materials theory, we are entering an era where the boundaries between simulation, prediction, and synthesis blur—and where the next great material discovery may come not from the lab, but from the algorithm.
PhD in Computational Physics, Head of R&D at Sber AI, Scientific consultant at AIRI. Alma mater: MIPT 2019, ESPCI 2014, NSU 2011.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in