
In 2012 the Liddle group reported a terminal uranium(V)-nitride, and a minor but significant part of that paper was that we showed the nitride could be protonated to form ammonia. The reason we undertook that protonation was to provide supporting evidence that the nitride was indeed a nitride, but also because uranium nitrides have long been implicated as intermediates in ammonia synthesis from when uranium was tested for use in the Haber Bosch process, where one of the open secrets since 1909 is that uranium is actually a better promoter of ammonia formation than iron.
When presenting our terminal uranium nitride chemistry at conferences, one question I have often been asked goes along the lines of: “so protons react with your nitride to give ammonia, but what about dihydrogen?” I have always felt that was a fair question, and usually answered [truthfully] that it was on the ever-expanding list of things to do just we hadn’t got around to doing it.
When considering reacting dihydrogen with our terminal uranium-nitrides, part of me thought it probably wouldn’t go anywhere since high oxidation state metal-nitrides are notoriously unreactive due to their very strong metal-nitrogen triple bonds, and only a handful have ever exhibited any chemistry with dihydrogen. However, on the other hand uranium often ‘breaks the rules’ so I thought it merited investigation, and it was becoming an itch that really had to be scratched. Fortunately, an exceedingly talented postdoc, Lucile Chatelain who is now an Associate Professor at the University of Brest, had just joined my group when this was on my mind so I thought it was a perfect project for her to tackle.
As we soon found out, it was synthetically an extraordinarily very challenging project, but with dogged tenacity and a real eye for detail Lucile made it work and carefully unpicked the whole story.
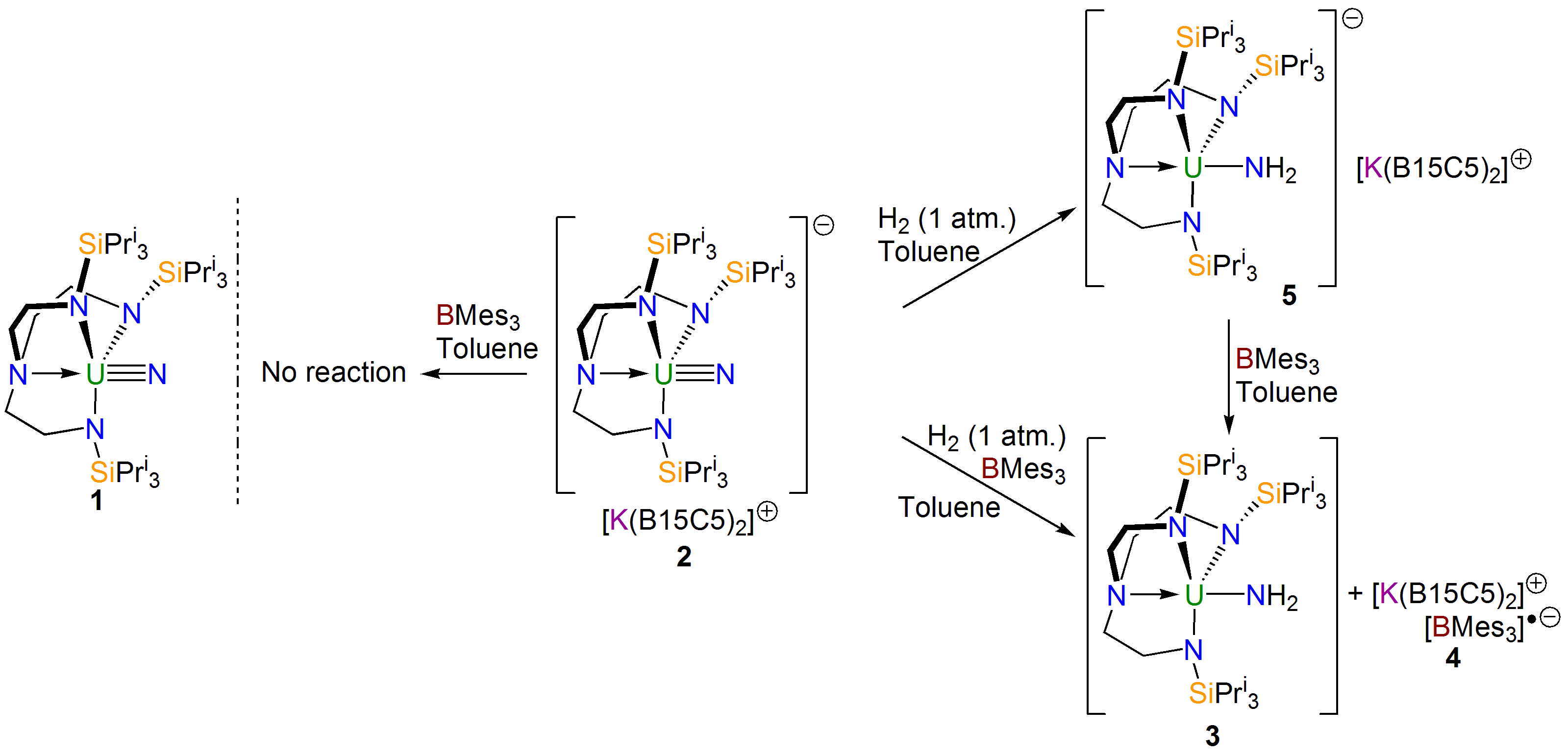
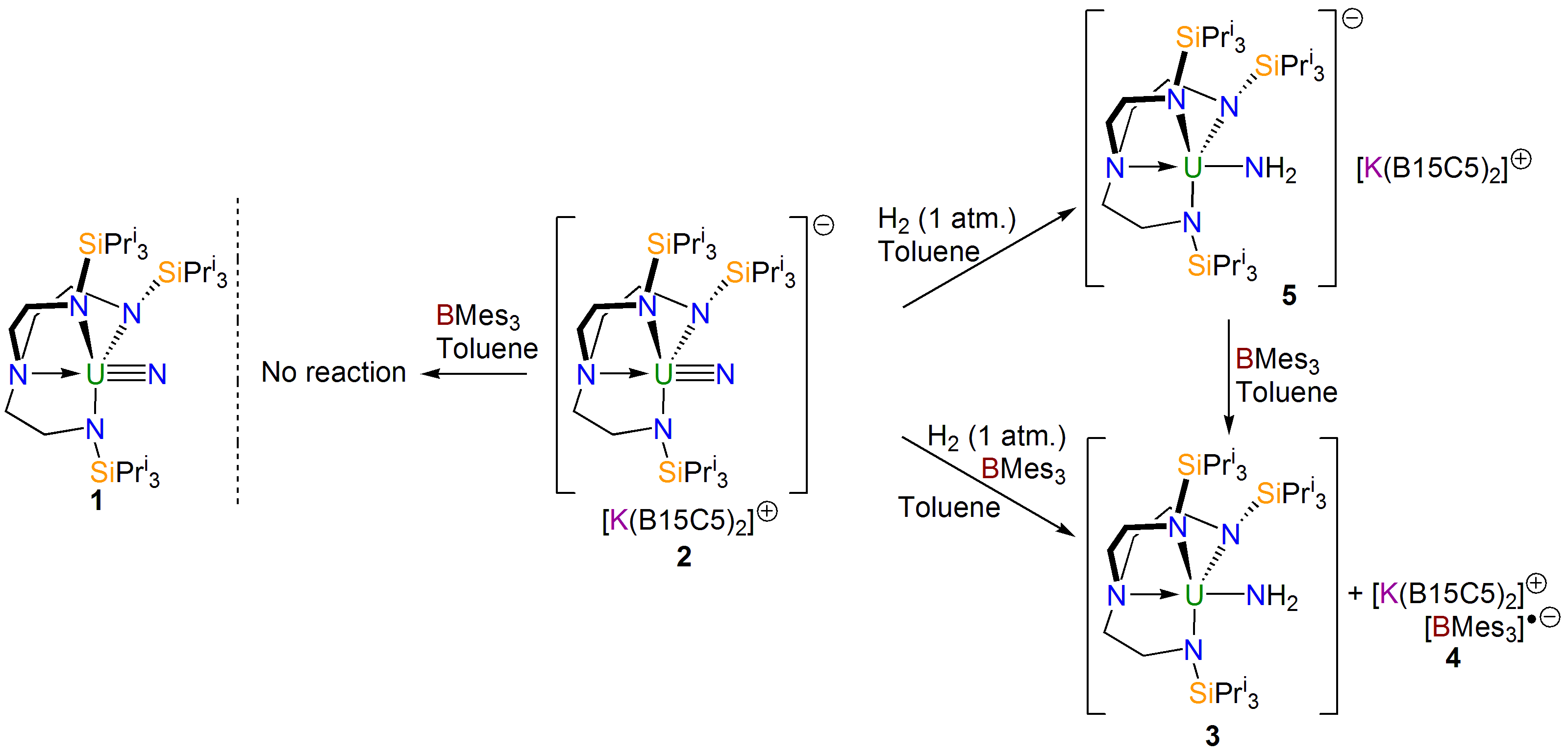
Quite quickly, we found that our uranium(VI)-nitride (1, see scheme above) really is completely unreactive towards dihydrogen, however change to a uranium(V)-nitride (2, see scheme above) and now some chemistry starts to happen. At first, so convinced were we that dihydrogen reactivity with these high oxidation state nitrides would be intrinsically disfavoured, we approached the problem using Frustrated Lewis Pairs (FLPs).
We found that mixing 2 and trimesitylborane, which has a Lewis acidic boron that is protected by the ortho-methyl groups of the mesityl rings, does not give any reaction. But, if dihydrogen was added to this mixture then we isolated a uranium(IV)-amide (3), resulting from hydrogenolysis of the nitride, along with a radical anion of trimesitylborane (4). We surmised that FLP chemistry was occurring, and also deduced that a uranium(III)-amide must be formed which then reduces the borane to give the uranium(V)-amide. To test this, we did away with the borane, though we thought we might kill the reactivity. However, to our surprise we were delighted to find that the hydrogenolysis still occurred, though now more slowly. Gratifyingly, sure enough we were able to isolate the predicted uranium(III)-amide (5), and could then oxidise it with borane to give 3 and 4.
With an understanding of the experimental outcomes of this reactivity, we enlisted the help of Laurent Maron’s group at the University of Toulouse to computationally model the work since we were keen to understand what was going on in these reactions.
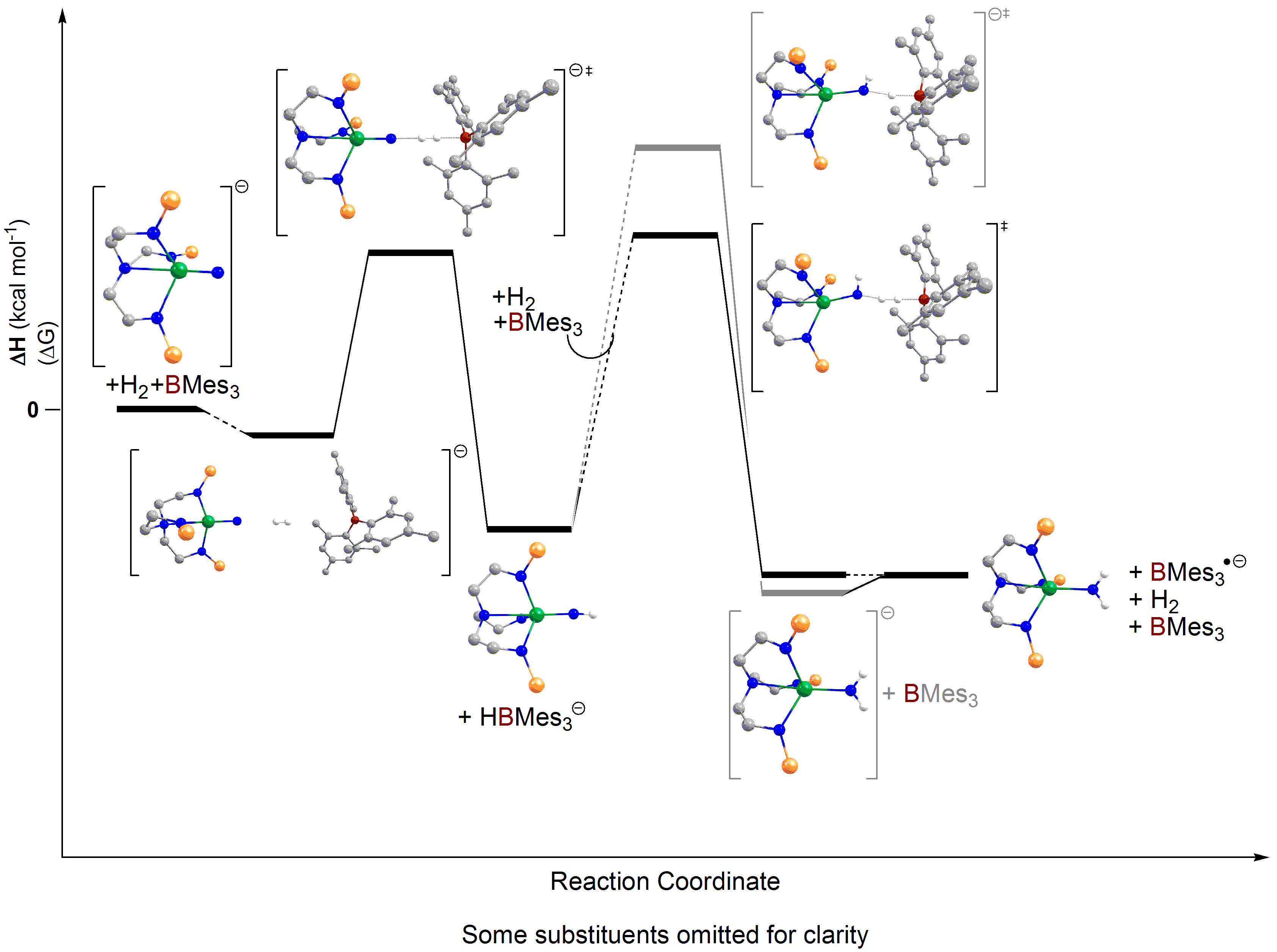
Laurent’s group was able to confirm that FLP chemistry indeed occurs when the borane is present in these hydrogenolysis reactions; the nitride is the Lewis base, the borane is the Lewis acid, and an encounter complex with dihydrogen occurs to give a heterolytic splitting of dihydrogen (see reaction profile above). It was then possible to find a second FLP dihydrogen heterolytic splitting event to deliver another proton atom to the nitride, nicely sidestepping the problem of how the hydride from the initial heterolytic splitting of dihydrogen could transfer to the nitride as a proton in a higher energy pathway.
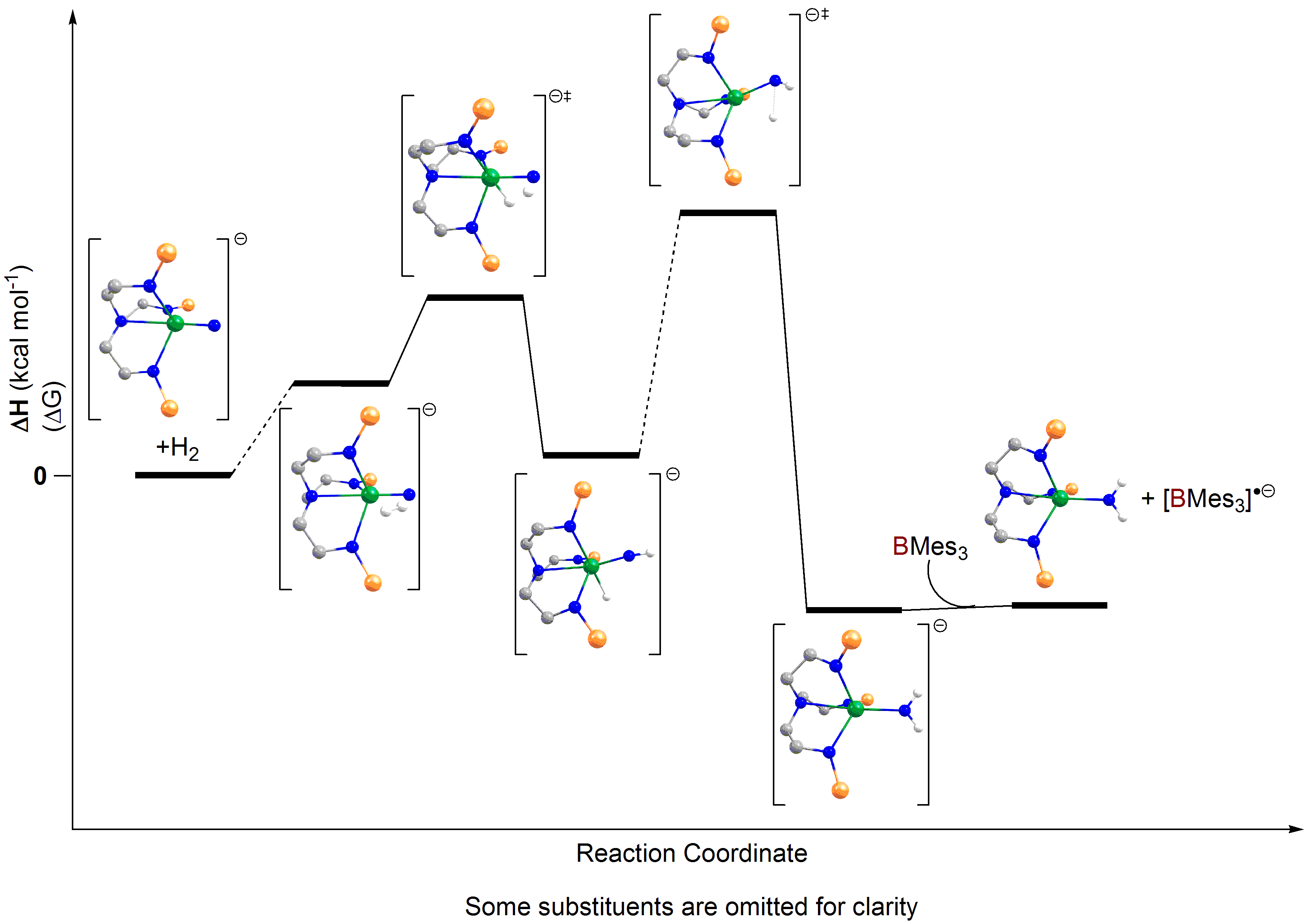
Laurent’s group were also able to compute the borane-free mechanism as well (see reaction profile immediately above), and found what amounts to direct dihydrogen addition across the uranium-nitride bond, but this is more difficult to achieve overall. These finding are in total agreement with the experimental result that the hydrogenolysis occurs faster with the borane than without, mainly because in this mechanism a hydride does have to transfer from uranium to nitrogen as a proton, concomitantly reducing the uranium.
So, what started out as an itch that needed scratching developed into a very demanding and detailed study with not one but two different types of metal-nitride hydrogenolysis mechanisms. The work introduced FLP chemistry to actinide science, revealed a high oxidation state terminal metal-nitride reacting with dihydrogen when any a priori assessment would predict no reactivity, and even though we weren’t expecting it may have revealed something else quite profound.
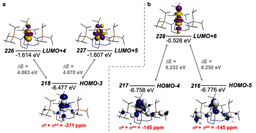
The fact that the uranium(VI)-nitride is inert to dihydrogen but the isostructural uranium(V)-nitride reacts is striking, since the extra valence electron in the latter is a 5f electron which would be normally thought of as non-bonding, so it shouldn’t precipitate such a profound change in reactivity. However, we’ve found in related uranium-nitride carbonylation chemistry with carbon monoxide that the extra 5f electron speeds up the carbonylation reactivity so a trend is emerging. From electronic structure studies on our terminal uranium(V)-nitrides we have found that the ligand field of the nitride is quite large at uranium, and the supposedly non-bonding 5f electron is clearly influenced by the nitride ligand field.
Now, the assertion that the supposedly non-bonding 5f electron is at the least not entirely non-bonding is looking to be backed up by reactivity data, which starts to open up all sorts of interesting questions and possibilities to consider about 5f electron participation in chemical bonding and reactivity.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in