Using phosphatase inhibitor beads coupled with mass spectrometry (PIB-MS) to parse phosphoprotein phosphatase signaling.
Published in Protocols & Methods

Kinase studies to understand phosphorylation signaling and identify new drug targets for multiple diseases are common, and with the development of multiplexed kinase inhibitor beads, more comprehensive than ever1. Equally important to phosphorylation signaling but largely understudied until recently is the reverse reaction of dephosphorylation by phosphatases. In our new Nature Protocol, we present phosphatase inhibitor beads (PIBs) as a tool to bridge the gap in knowledge between kinase regulation of phosphorylation and phosphatase regulation of dephospohrylation.
Protein phosphorylation is the reversible addition of a phosphate group onto a serine, threonine, or tyrosine residue. The phosphorylation reaction is catalyzed by kinases and reversed by phosphatases. The kinase to phosphatase ratio for tyrosine residues is near 1:1, but for serine and threonine residues, the ratio drastically increases: while approximately 400 kinases phosphorylate serine and threonine residues, over 90% of the corresponding dephosphorylation reactions occur through the activity of a small family of 7 catalytic subunits and their interacting proteins, the phosphoprotein phosphatase (PPP) family. This radical difference in unique proteins led to the belief that phosphatases were unspecific, but more recently it has been revealed that PPPs obtain specificity through interactions with numerous scaffolding and regulatory subunits (PPPome)2. As the most abundant type of post translational modification3, phosphorylation plays important roles in countless signaling pathways including cell growth and division4, and deregulated kinase and phosphatase signaling has been implicated in many diseases such as cancer2. To combat this, numerous studies have focused their attention on kinases, identifying substrates and interaction networks and developing many successful drugs for disease treatment1. Unfortunately, many cancers treated with kinase inhibitors develop resistance, and new strategies for treatment need to be discovered. For this purpose, we turned from kinases and phosphorylation to phosphatases and dephosphorylation. We developed PIB-MS to understand the endogenous dephosphorylation signaling network and how to leverage it for disease treatment.
PIB-MS is an ideal strategy to systematically identify and study the PPPome. PIBs are comprised of the PPP inhibitor MCLR covalently bound to sepharose beads. MCLR binds to 5 of the 7 PPP catalytic subunits with high affinity and, when bound to beads, enable the capture and identification of scaffolding, regulatory, and interacting proteins. Due to the conserved nature of PPPs, PIBs can be used with cells from yeast to humans, and are compatible with small samples sizes, such as clinical samples2.
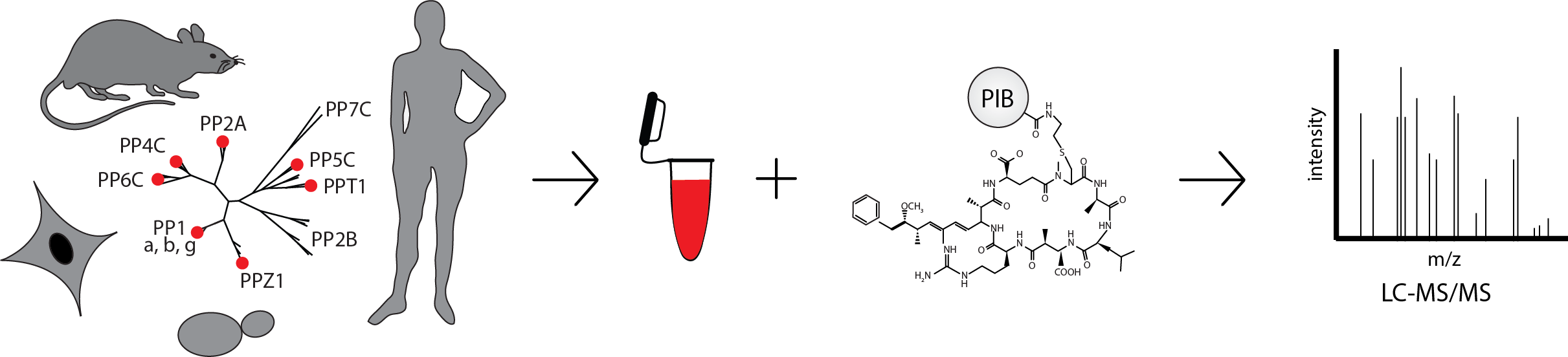
So far, PIBs enabled the discovery of new PPP interacting proteins and identification of cell-, tissue-, or species-specific PPP expression patterns. We have used this list to profile expression differences between brain and breast cancer cell lines and between several different mouse tissues2. Using PIBs in this manner, we should be able to comprehensively map PPP expression in all human tissues and get a better understanding of the roles each PPP plays in each tissue and how those roles are perturbed in cancer and other disease. More recently, we investigated the regulation of PPPs by phosphorylation and discovered a phosphorylation site on the catalytic subunit of PP2A that is responsible for driving mitotic entry4. Experiments such as this that study post translational modifications will add another layer of understanding to how PPPs are regulated in normal and disease tissues and hopefully open new avenues for treatment.
In our Nature Protocol, we introduce PIBs and provide a step-by-step protocol from cell lysis through PIB affinity purification, TMT labeling, phosphorylation enrichment, and data analysis, giving examples of when and why each application would be used. The protocol is easy to follow, provides troubleshooting help, and discusses expected results and how to analyze them. Users should be able to successfully master PIB-MS with this protocol.
Our hope is for PIB-MS to facilitate understanding of phosphoprotein phosphatases on a systematic level, to learn how they are regulated and what signaling processes they themselves regulate, and to one day be able to use this knowledge towards new and better disease treatment.
References
- Collins, Kyla A L et al. “Proteomic analysis defines kinase taxonomies specific for subtypes of breast cancer.” Oncotarget vol. 9,21 15480-15497. 29 Jan. 2018, doi:10.18632/oncotarget.24337
- Lyons, Scott P et al. “A Quantitative Chemical Proteomic Strategy for Profiling Phosphoprotein Phosphatases from Yeast to Humans.” Molecular & cellular proteomics : MCP vol. 17,12 (2018): 2448-2461. doi:10.1074/mcp.RA118.000822
- Khoury, George A et al. “Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database.” Scientific reports vol. 1 (2011): 90. doi:10.1038/srep00090
- Nasa, Isha et al. “Quantitative kinase and phosphatase profiling reveal that CDK1 phosphorylates PP2Ac to promote mitotic entry.” Science signaling vol. 13,648 eaba7823. 8 Sep. 2020, doi:10.1126/scisignal.aba7823
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in