VESPA: Unlocking the covert pathways of resistance to targeted cancer therapies with machine learning
Published in Cancer, Protocols & Methods, and Cell & Molecular Biology
Colorectal cancer (CRC) accounts for about 10% of cancer-related deaths. While targeted drugs have emerged as a leading therapeutic approach in recent years, their effectiveness is invariably affected by the emergence of cell-adaptive resistance mechanisms. The rewiring and adaptive response of signaling networks may play a pivotal role in this phenomenon. However, the intricate and poorly understood nature of signal transduction pathways involved in these processes complicates our systematic understanding of adaptive drug resistance.
Embarking on this challenge felt like the perfect opportunity for my postdoctoral research within the Califano group at Columbia University. Our objectives were clear yet somewhat ambitious; specifically we wanted to: (a) Establish a representative model system for clinical CRC subtypes suitable for drug screening, (b) Generate phosphoproteomic profiles representing the time-dependent response of these models to large-scale drug perturbations, (c) Develop an algorithm to reverse engineer signaling networks architecture in CRC by data-driven machine learning-based analysis of these time series, and (d) Develop an additional algorithm to interrogate these networks to identify the key proteins that may mediate the cell’s adaptive response. We ended up calling these algorithms dVESPA and mVESPA (Virtual Enrichment-based Signaling Protein-activity Analysis), respectively.
, evaluates activity at phosphostate- and activity-levels (b), and distinguishes between direct and indirect interactions. At the activity-level, abstract \"activation/deactivation\" events better associate targets for kinases and phosphatases.")
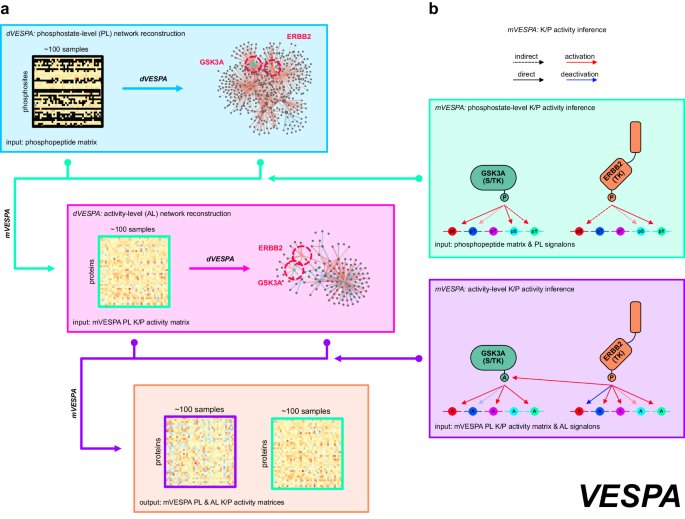
Figure 1: VESPA assesses protein kinase and phosphatase activity based on substrate phosphostate. Input is a matrix of phosphopeptide abundance across conditions. The method reconstructs signaling networks, generates signalons for each enzyme (a), evaluates activity at phosphostate- and activity-levels (b), and distinguishes between direct and indirect interactions. At the activity-level, abstract "activation/deactivation" events better associate targets for kinases and phosphatases.
Choosing suitable model systems for perturbational profiling was in itself a complex process. Leveraging MOMA (Multi Omic Master Regulator Analysis), an algorithm developed by the Califano group, we identified six colorectal cell lines from the Cancer Cell Line Encyclopedia (CCLE) that represented the diverse clinical subtypes of colorectal cancer and were also relatively easy to culture in vitro. Briefly, MOMA stratifies tumor subtypes based on the activity of Master Regulator (MR) proteins that canalize the effect of upstream genetic alterations to implement a specific transcriptional cell state. We thus attempted to find cell lines that would recapitulate the MR-based subtypes identified by MOMA, under the assumption that these may also be associated with distinct signaling network activity, as confirmed by subsequent analyses.
The next step involved selection of a targeted drug panel (7 drugs plus 1 DMSO control) targeting distinct signaling pathways in CRC and potentially clinically relevant. The panel was used to perturb the six CRC celllines selected by MOMA analysis, across eight time points, spanning from 5 minutes to 96 hours. Crucially, we ensured that the concentrations of the drugs were kept well below levels that would completely inhibit the targets or induce cell death. Our primary focus was on observing the adaptive responses that arise following sustained perturbation of the signaling pathways involved. Phosphoproteomic profiles were then generated using a label-free data-independent acquisition protocol and the IPF (Inference of PeptidoForms) algorithm, previously developed with Yansheng Liu (Yale University) during our joint time in Ruedi Aebersold's Lab at ETH Zurich. Samples were then profiled at multiple time points following pharmacologic perturbation in 354 individual Mass Spec runs. This meticulous approach yielded data characterized by an outstanding balance between coverage and quantitative consistency, a feat virtually unattainable with other methodologies.
Once these data were available, the development of the two VESPA algorithms emerged as the focal point of our project (Figure 1). Initially, we considered applying the established ARACNe and VIPER algorithms to phosphoproteomic profiles, without modifications. However, we quickly realized that a proteomics-specific version of these algorithms was required because the results produced by their native implementation were poor. Specifically, we extended ARACNe to handle sparse data matrices and modified its network pruning step to align with kinase and phosphatase mechanism specificity. Additionally, characterization of proteins with poorly measurable substrates, such as tyrosine kinases, required implementation of a two-step hierarchical approach (Figure 2). Taken together, These changes helped us assemble accurate and comprehensive disease-specific signaling networks de novo, from large-scale phosphoproteomic profiles of clinical samples. The resulting VESPA-inferred CRC signaling network comprised seven times more enzymes/substrates interactions than Pathway Commons. We speculated that this enhanced coverage, coupled with specificity from network pruning, could facilitate cross-talk correction during kinase or phosphatase activity inference. Quantitative benchmarks supported this hypothesis, showing that VESPA significantly outperforms previously published methods, especially for context-specific studies.

Figure 2: Illustration of the comparison between VESPA-inferred kinase activities and measured phosphopeptides, highlighting VESPA's enhanced sensitivity, particularly in the absence of directly measured tyrosine-phosphopeptides.
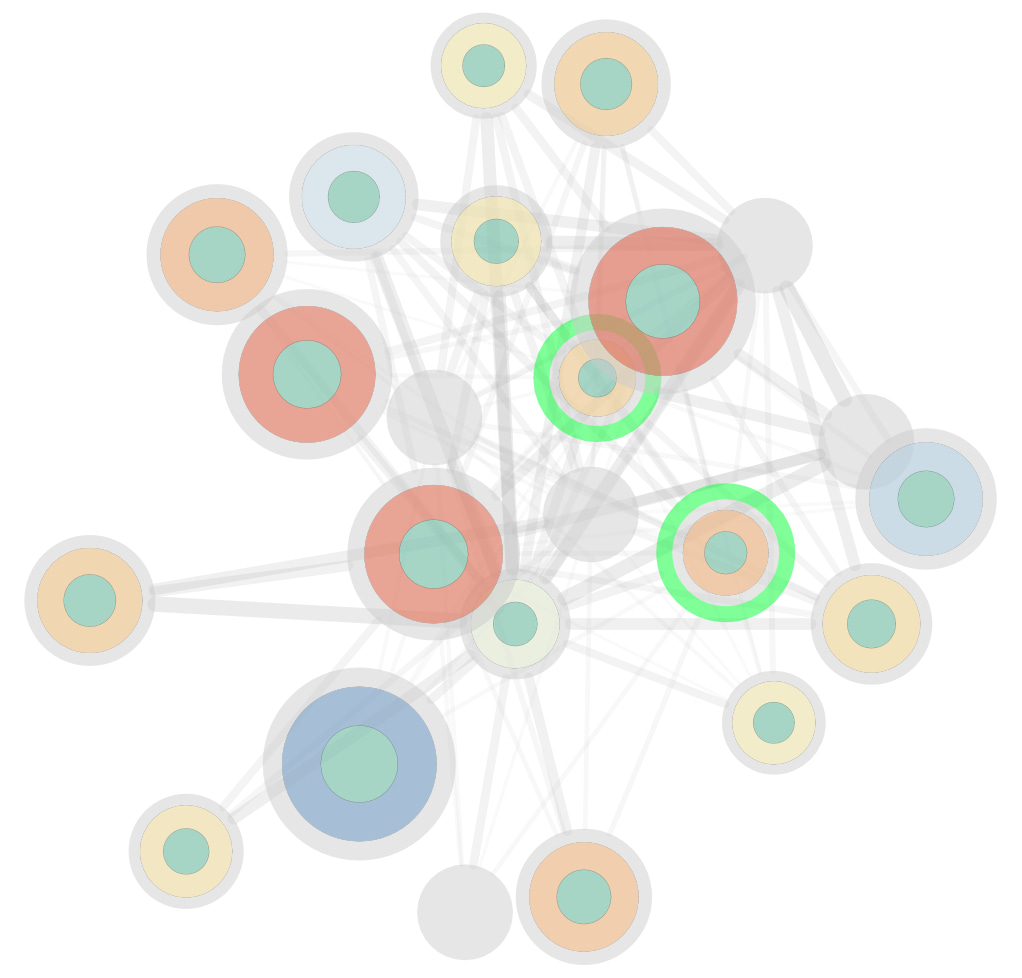
With all components now in place, our initial aim was to evaluate how signaling rewiring impacts kinase and phosphatase activity. To accomplish this, we utilized the DeMAND (Detecting Mechanism of Action by Network Dysregulation) algorithm to assess network dysregulation. This algorithm pinpointed the distinct signaling interactions primarily affected by drug perturbation. For instance, our analysis revealed that while osimertinib perturbation in HCT-15 and HT115 initially inhibited a similar set of targets, adaptive responses led to divergent activation of compensating mechanisms at later time points (see Figure 3). This example underscores the importance of considering the drug's mechanism of action within specific cellular contexts.
 of the EGFR inhibitor osimertinib. Nodes represent highly affected regulators, with inner circle colors indicating cell line type and outer circle color and size indicating VESPA activity. The legend for VESPA activity is shown. Edges denote dysregulated, undirected interactions between KP-enzymes, with line thickness reflecting statistical significance. Proteins highlighted in green are known primary/secondary targets.")
Figure 3: The figure depicts network dysregulation and the mechanism of action (MoA) of the EGFR inhibitor osimertinib. Nodes represent highly affected regulators, with inner circle colors indicating cell line type and outer circle color and size indicating VESPA activity. The legend for VESPA activity is shown. Edges denote dysregulated, undirected interactions between KP-enzymes, with line thickness reflecting statistical significance. Proteins highlighted in green are known primary/secondary targets.
In a final step, we expanded our analysis to identify candidate "resistance factors", namely kinase/phosphatase enzymes activated in adaptive responses to drug perturbation. By comparing late versus early perturbation time points, we generated a candidate list for each cell line and drug perturbation. While many of these candidate proteins had been previously associated with colorectal tumorigenesis and/or drug resistance in the literature, our ultimate objective was to experimentally validate them in systematic fashion. For this purpose, we assessed cell line-specific changes in drug sensitivity following pooled CRISPR knock-out of all kinases and phosphatases. VESPA showed remarkable predictive power, yielding AUROC values of 0.81 and 0.74 for HCT-15 cells treated with linsitinib and trametinib, respectively, thus validating its ability to support context-specific, systems-wide elucidation of signaling networks and cell-adaptive drug response.
In summary, VESPA stands out as a significant advancement in the realm of kinase activity inference and signaling network reverse engineering algorithms. What sets it apart are its versatile features applicable to a wide range of experimental designs. From its machine learning-driven, context-specific signaling network generation to its capability for cross-talk correction and hierarchical activity inference, VESPA offers unprecedented resolution at the phosphosite level. Our study confirmed VESPA as a state of the art network reverse engineering and interrogation algorithm. Given the wealth of large-scale clinical phosphoproteomic profiles available from initiatives like CPTAC, VESPA represents a key addition to the cancer research toolset, especially in terms of the signaling network contributions to drug sensitivity.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in