When Less is More (Dangerous): How Centrosome Loss Drives Chromosomal Instability?
Published in Cancer, Cell & Molecular Biology, and Genetics & Genomics
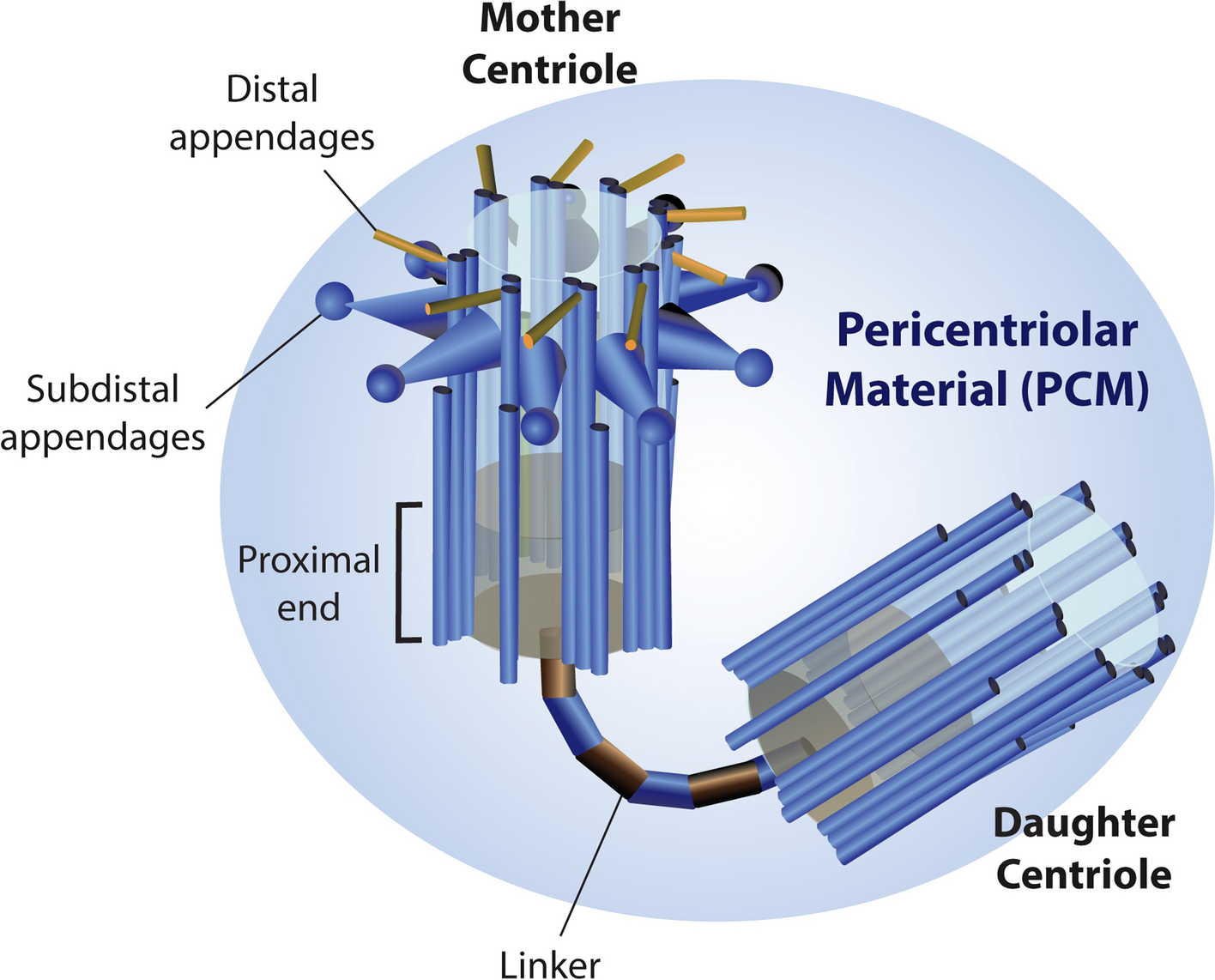
For decades, cancer researchers have looked at tumor cells and observed too many centrosomes.
In 1914, the German cytologist Theodor Boveri hypothesized that centrosome aberrations could contribute to human cancer. Indeed, centrosome defects, especially amplification (the production of supernumerary centrosomes) are present in a broad array of solid and hematopoietic human cancers. They correlate with advanced tumor grade and poor clinical outcomes. At present, centrosome amplification is recognized as an emerging hallmark of cancer cells, known to drive multipolar spindle formation and tumor aggression.
But is the opposite scenario - having fewer or no centrosomes - equally dangerous?
One of the most frequent questions we were asked when presenting our study that explores the impact of centrosome loss is: If a cell is missing this critical organelle, can it still divide? The answer is yes. Cells do not simply stop dividing; instead, they divide and make mistakes when doing so. Loss of the centrosomes forces a cell to rely on “Plan B”, an acentrosomal spindle assembly pathway prone to errors, leading to chromosome segregation defects that cause a massive chaos to the cell's genome. In most healthy cells, centrosome loss results in robust cell cycle arrest within a few divisions via a pathway that requires p53 – the famous guardian of the genome. However, there are "evaders". These rare cells escape cell cycle arrest and, instead, potentially evolve into aggressive malignancies.
Connecting the Organelle to the Disease
Prostate cancer differs from many other malignancies. It harbors few point mutations in oncogenic driver genes and instead shows extensive structural variations and gene fusions induced by rearrangements – a cancer hallmark known as chromosomal instability (CIN). The consequences of centrosome loss have long piqued our lab's interest. In a previous study published in Oncogene (2019), our colleague Dr. Mengdie Wang first discovered centrosome loss in human primary prostate cancer tissue. This phenomenon was more prevalent than expected and correlated with tumor grade. In subsequent in vitro and in vivo models, we isolated clones that had experienced transient centrosome loss. These cells not only survived the process but, as a result, some were also transformed. This led us to ask - what are the genomic alterations that transformed these cells?
From Chaos to Signatures
To understand the molecular alterations underlying centrosome loss-induced tumorigenicity, we initiated our current study. We performed whole-genome sequencing on our cell and tumor models. The data confirmed that cells resulting from transient centrosome loss displayed extensive CIN. Using the CIN genome generated from our experimental model, we analyzed large cohorts of prostate cancer patient samples. We identified recurrent genomic alterations in patient samples that clustered closely with our transient centrosome loss models. Furthermore, by integrating genomic classification with transcriptomic data, we identified a signature of 9 CIN-associated genes, which we termed CIN9. This transcriptomic signature represents either a cause or a consequence of cells evading from checkpoints and surviving the resulting genomic chaos from centrosome loss.
The "Hopeful Monster"
Our experimental model was designed to cause a burst of genomic crises by transiently removing centrosomes in cultured cells, similar to that observed in prostate cancer. Interestingly, our model produced a clone that continued to display CIN, eventually giving rise to a tumorigenic subclone that aggressively formed xenograft tumors in mice within just three weeks. Our findings suggest that a cell with CIN acts like the evolutionary "hopeful monster", an organism with profound ability to fit an environment perfectly and sweep the population. Understanding these CIN-associated features may help us differentiate between benign and aggressive tumors and eventually lead to precision treatment strategies.
Continuous questions about centrosome loss in cancer
Centrosome loss appears to be a primary cause of CIN in prostate cancer. But what leads to the loss of centrosomes in the epithelial cells of prostate glands in the first place? That is the next question we intend to answer.
Follow the Topic
-
Cellular and Molecular Life Sciences
Cellular and Molecular Life Sciences (CMLS) is a multidisciplinary Open Access journal covering the latest aspects of biological and biomedical research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in