Physics-Based Molecular Dynamics Simulations Shed Light on Protein-Ligand Interactions
Published in Chemistry and Cell & Molecular Biology
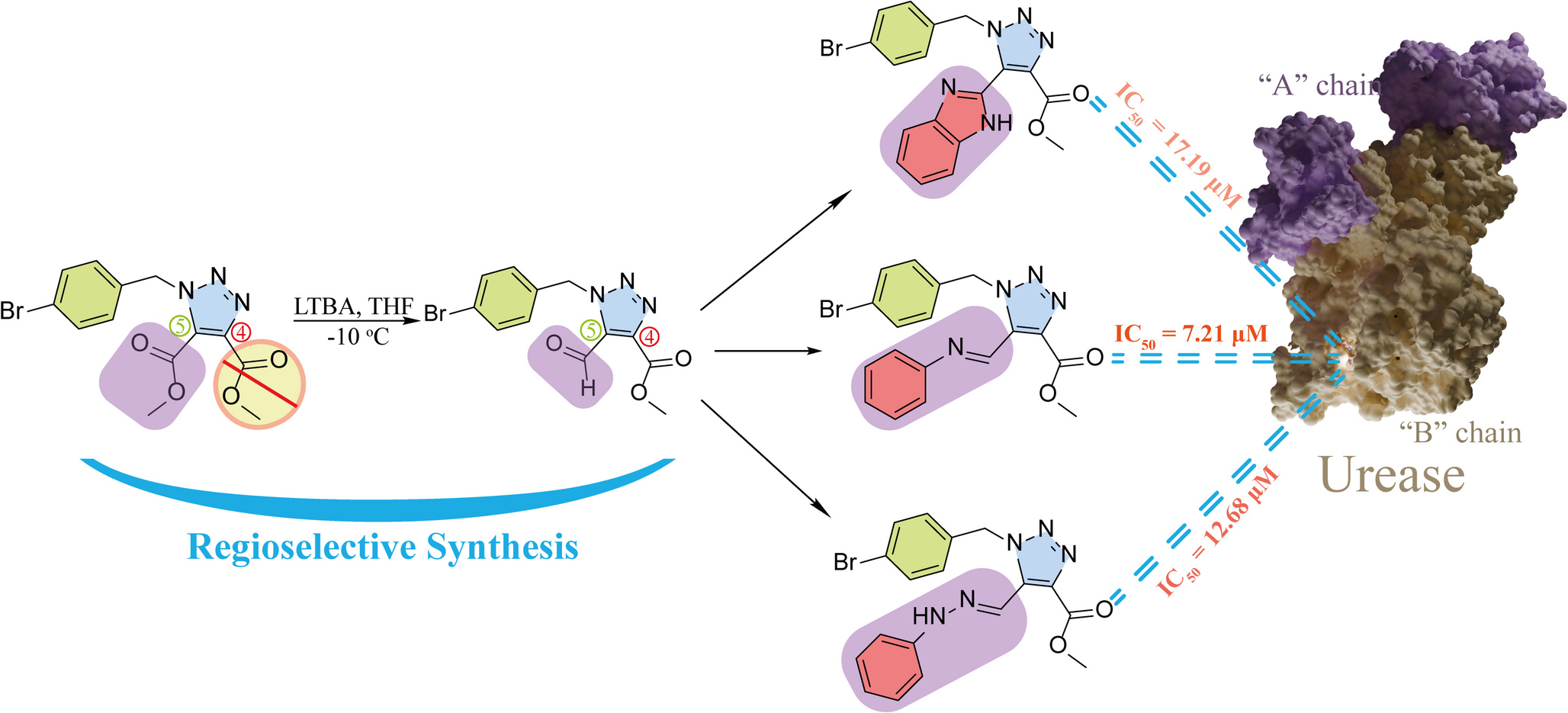
This video contains a similar study to the one featured in the article on the Novel 4-Bromobenzyl-1H-1,2,3-Triazole Scaffold. The article describes the broad-spectrum multi-enzyme inhibition of newly discovered potent urease inhibitors, which were tested for protein-ligand complex stability using molecular dynamics simulations.
Molecular dynamics simulations also allow important conclusions to be drawn about the functioning of protein dynamics. Stable conformations can be determined by applying dimensional reduction to the continuously updated atomic position data obtained from molecular dynamics simulation data. The most important situations for this are captured by dimensional reduction algorithms.
The article Novel 4-Bromobenzyl-1H-1,2,3-Triazole Scaffold takes this a step further and integrates dimensional reduction with MM/GBSA free energy calculations. This has resulted in the following image:

Looking at the image, it can be seen that a similarity matrix forms a cluster by displaying similar protein conformations together. The MM/GBSA integration shows this cluster as a community consisting of structures with very low energy. This is because the free energy calculation added to the graph as the third dimension is scaled by colours. In addition, a higher-energy and less dense community indicates metastable states. The high-energy, i.e. unstable conformations that form the first frames of the simulation are seen in the graph as a broad scattering area extending upwards and indicate high-energy states in the energy values calculated throughout the simulation.
You can also analyse your simulation data in depth by visiting the comprehensive GitHub repository on dimensional reduction in molecular dynamics simulations.
For more molecular dynamics videos, you can visit the ortaakarsu.net website and the PharmSciPulse YouTube channel.
A. Buğra Ortaakarsu is a medicinal chemist and researcher with a strong focus on molecular modeling, particularly in molecular dynamics, protein dynamics, quantum chemistry, and density functional theory (DFT). As a member of Sigma Xi, The Scientific Research Honor Society, he is committed to advancing scientific knowledge through the rigorous investigation of chemical–protein interactions, with the goal of developing precise, physically grounded, and innovative analytical methodologies.
His research includes the study “Synthesis, Design, and Cholinesterase Inhibitory Activity of Novel 1,2,4-Triazole Schiff Bases: A Combined Experimental and Computational Approach,” which aims to identify promising therapeutic candidates for Alzheimer’s disease and other neurodegenerative disorders by integrating experimental findings with advanced computational techniques.
Adopting a highly interdisciplinary perspective, Ortaakarsu combines advanced mathematics, game theory, Riemannian geometry, and differential geometry to address complex scientific and strategic problems. By embedding game-theoretical principles into artificial intelligence frameworks, he develops optimized, adaptive systems applicable across diverse scientific and technological domains.
Driven by a strong commitment to scientific rigor and innovation, Ortaakarsu continues to design and refine novel methodologies at the intersection of medicinal chemistry, advanced geometry, computational modeling, quantum chemistry, and DFT, with a clear emphasis on translating theoretical insights into robust, real-world applications.
Follow the Topic
-
Journal of Pharmaceutical Innovation
This is a multidisciplinary, peer-reviewed scientific journal focusing on drug development, manufacturing, process control, and technology.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in