A fluorine-thiol displacement reaction as a peptide stapling platform
Published in Chemistry

Targeting protein-protein interactions (PPIs) has become increasingly attractive due to their dysregulation which is frequently associated with the development of cancers and neurodegenerative disease1. Conventional small molecule based approaches aimed at targeting PPIs have remained unsuccessful owing to the flat and shallow nature of α-helices commonly found at protein interfaces2. One alternative method employs the use of chemical stapling to lock peptides in a bioactive α-helical secondary structure. Current crosslinking methods such as ring-closing metathesis (RCM) have produced helical peptides that exhibit high specificity, improved binding affinities, and proteolytic stability. Furthermore, these peptides have the potential to serve as molecular probes or as future therapeutics to target PPIs3. However, these approaches suffer from limited aqueous solubility resulting from their hydrophobic linkers and more importantly suffer from poor cell permeability like most other macromolecules4.
Recently, our group developed a novel bio-orthogonal reaction that can selectively displace fluorine substituents alpha to amide bonds in the presence of a benzenethiol under mild basic conditions5. The reaction termed, "fluorine-thiol displacement reaction" (FTDR), displayed second-order reaction rate kinetics which is comparable to the Staudinger ligation. Given the bio-orthogonal nature of the fluorine-thiol displacement reaction, the platform was previously used to label both histone and non-histone acetylation substrates. A prior study had demonstrated that the thiol side chain of cysteine residues within a polypeptide could react with a proximal fluoroacetamide group6. This inspired us to utilize the efficient and facile FTDR platform towards the development of crosslinked peptides.
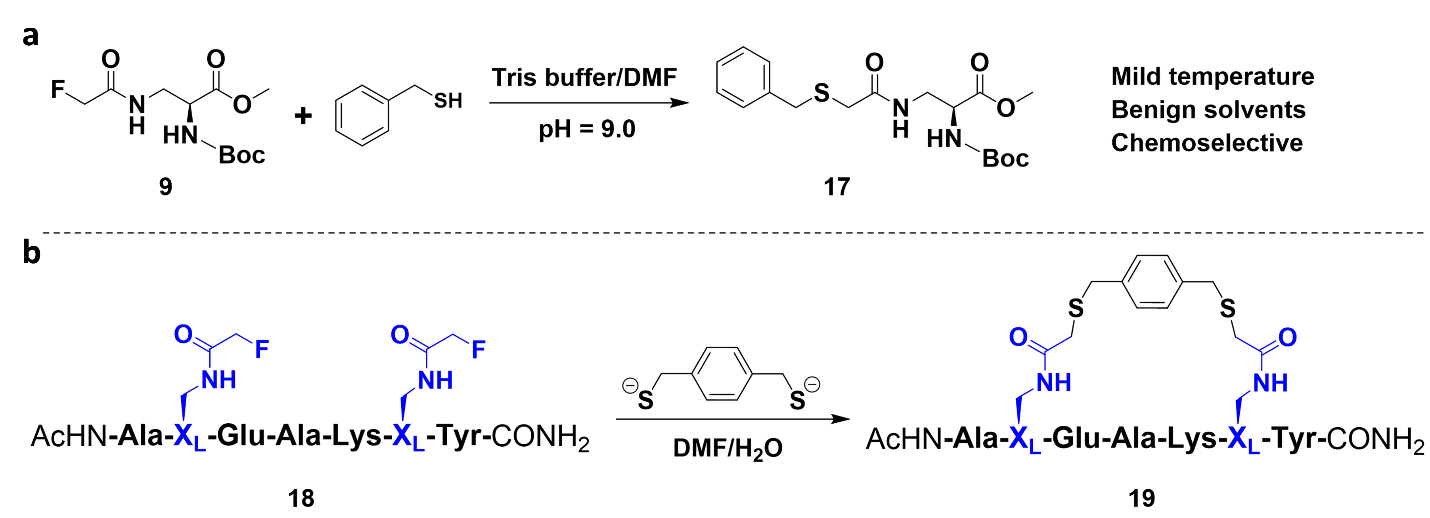
Fig. 1 Model fluorine-thiol displacement reactions. (a) Model reaction between the α-fluoroacetamide building block and a benzyl thiol. (b) Macrocyclization of an unprotected model peptide with a 1,4-benzenedimethanethiol linker using the FTDR platform. XL denotes the α-fluoroacetamide highlighted in blue.
We first set out to evaluate the reaction of the reported fluoroacetamide compound with various nucleophiles such as hydrazine, cysteine, and other thiol containing compounds in mildly basic Tris (tris(hydroxymethyl)aminomethane conditions. Delightfully, the fluoroacetamide compound displayed a strong preference for thiols over other nucleophilic functional groups. At pH 9.0-9.5, benzyl thiol enhanced the conversion of fluoroacetamide much better than cysteine, potentially due to the fluorine-π interaction. Encouraged by this, a model protected fluoroacetamide building block 9 was synthesized consisting of a natural amino acid backbone bearing a fluoroacetamide side chain (Fig.1a). Under FTDR conditions, compound 9 was efficiently converted to compound 17 in good yield. Interested in the chemoselectivity of our FTDR methodology and its efficacy as a macrocyclization strategy we synthesized peptide 18 which possesses multiple unprotected functional groups in addition to α-fluoroacetamide building blocks placed in an i, i+4 stapling fashion (labeled XL highlighted in blue) (Fig.1a). Peptide 18 was subjected to FTDR conditions in the presence of the benzyl thiol cross linker 1,4-benzenedimethanethiol and the conversion to the macrocyclized product 19 was greater than 90% after 12 h (Fig.1b).
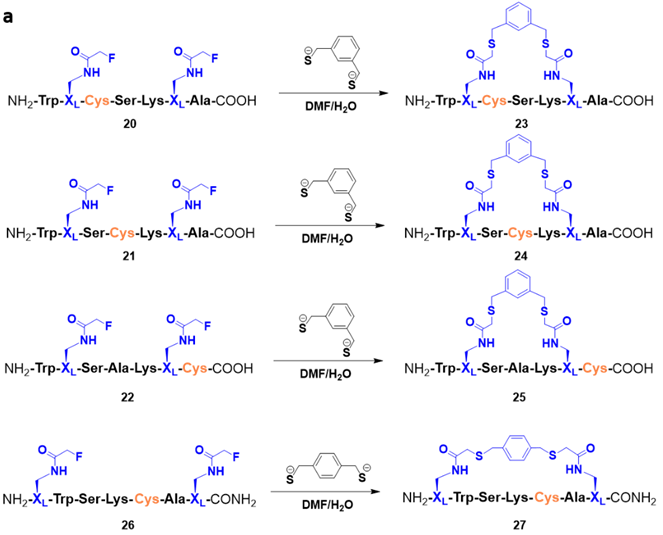
Fig. 2 FTDR macrocyclization of cysteine-containing peptides with cysteine and XL at different positions.
To further investigate the chemoselectivity of the FTDR macrocyclization platform we decided to synthesize model peptides containing additional unprotected functional group chemistries. Particularly, we were interested to see if the unprotected cysteine could be spared in the reaction given the preference of α-fluoroacetamide for benzyl thiols (Fig.2). The fluorine-displaced macrocyclized major product was observed for all model peptides 23-25 and 27, and the conversion was also greater than 90% after 12 h. These results suggest that the FTDR platform is both chemoselective and bio-orthogonal to various canonical functional groups present on amino acid side chains including the sulfhydryl group of cysteines.
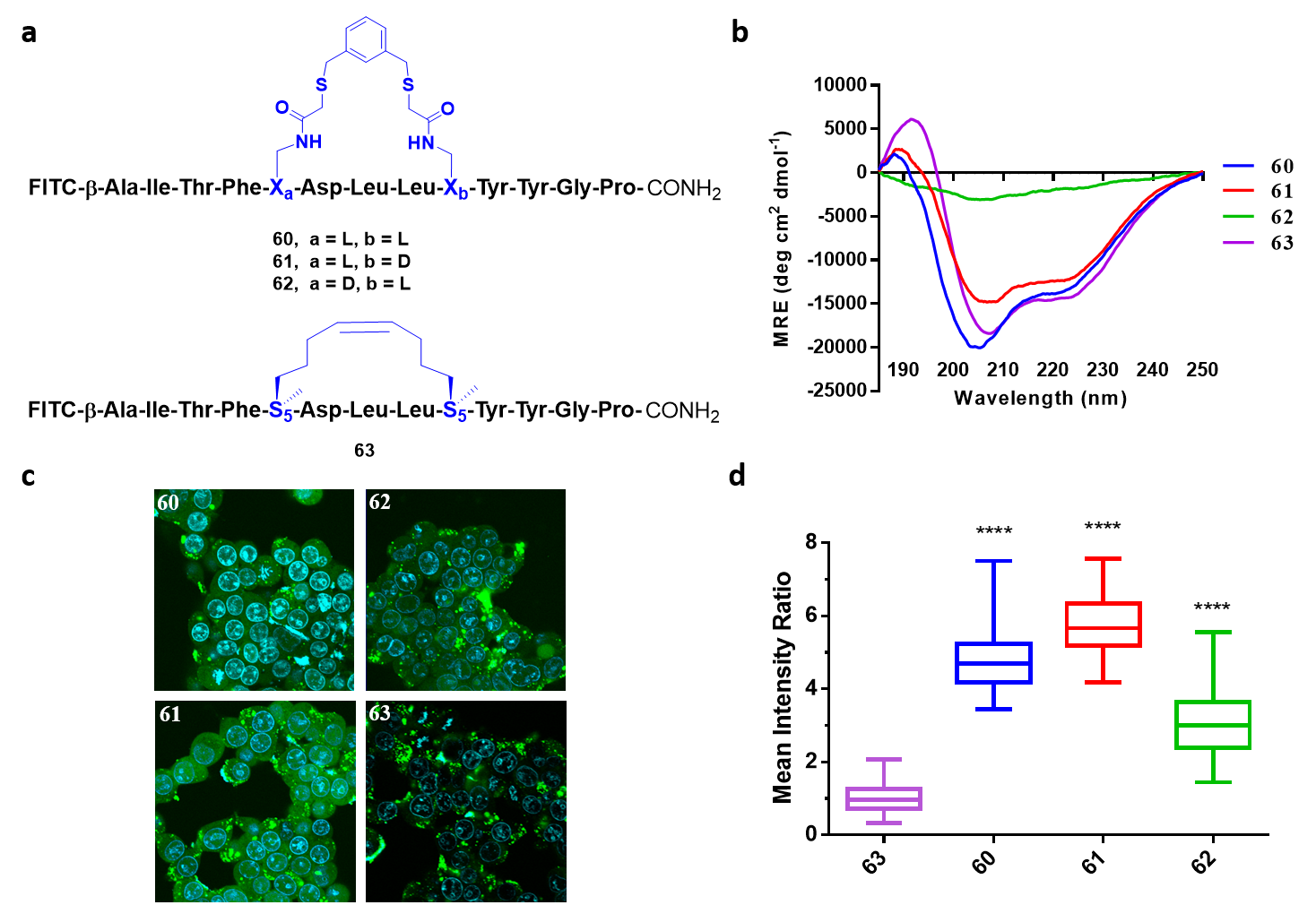
Fig. 3 FTDR based stapled HIV C-CA analog. (a) Model FTDR HIV C-CA stapled peptides 60-62, and peptide 63 a previously reported RCM based stapled peptide. (b) CD spectra of peptides 60-63. (c) Fluorescent confocal microscopy images of HEK293T cells upon treatment with peptides 60-63. Blue color is the nucleus stain using Hoechst 33342; Green color is the FITC-labeled peptides. (d) Quantification of cell penetration for peptides 60-63. “****” Represents p < 0.0001.
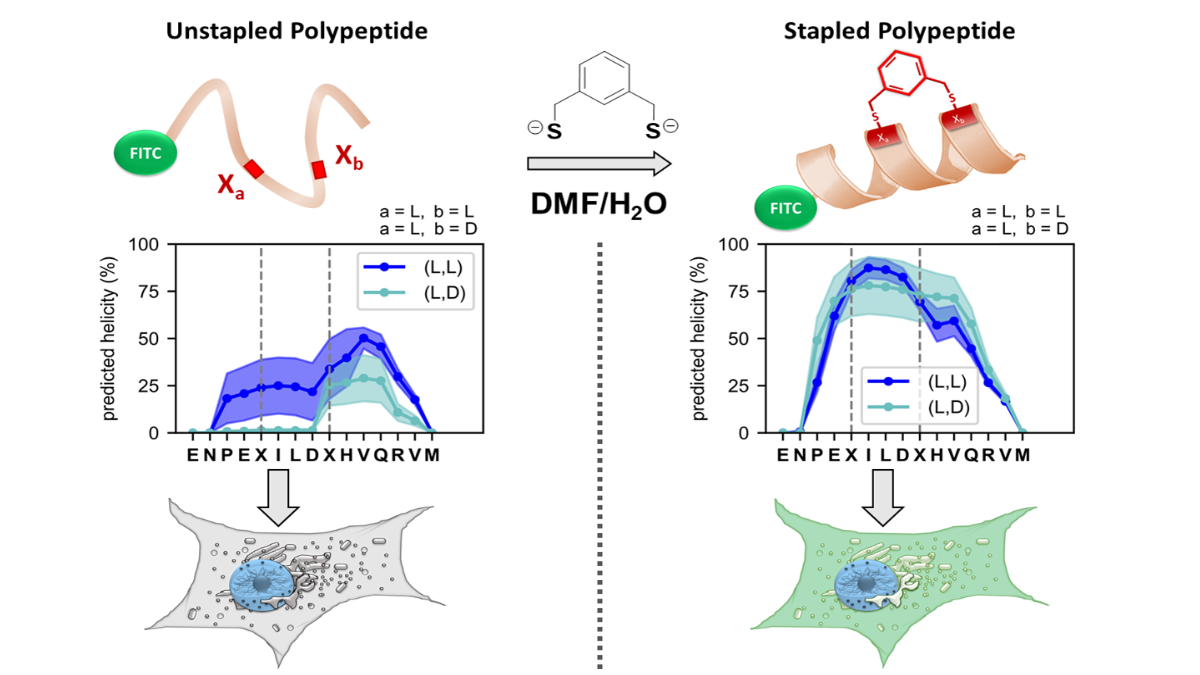
Following our investigation into the chemoselectivity of the reaction we next wanted to explore the chirality preference for various FTDR based stapled peptides. One model peptide, HIV C-CA was previously shown to exhibit a stable α-helical secondary structure following traditional RCM based i, i+4 stapling. To test the chirality preference of our FTDR approach we synthesized model HIV C-CA peptides 60-62 containing different α-fluoroacetamide substrates bearing different chirality combinations, and compared their α-helical character to the reported RCM based peptide 63 (Fig.3a). The CD (Circular Dichroism) spectral data showed that model HIV C-CA peptides 60 (L,L) and 61 (L,D) exhibited comparable helicity to the RCM control 63, whereas peptide 62 (D,L) showed minimal helicity possibly owing to linker strain (Fig. 3b). With confirmed α-helical peptides 60 and 61 in hand, we then tested their cell permeability since peptide 63 previously showed enhanced cellular uptake following the stabilization of the α-helical secondary structure using RCM. All peptides were FITC-conjugated to allow for their intracellular visualization and subsequent quantification. HEK293T cells were treated with peptides 60-63, and a stronger fluorescence signal correlating with a higher cellular uptake was seen for the FTDR peptides 60-62 compared to the RCM control 63 (Fig 3c, d). Interestingly, the higher uptake of the FTDR based peptides 60-62 suggests that the aromatic moiety of the linker may have enhanced the cellular uptake. Enhanced cellular uptake was also observed with other model peptides such as Axin, STAD, and SHABA peptides. Preliminary mechanistic studies suggested that cell penetration occurred through multiple endocytosis pathways, a phenomenon distinct from traditional RCM peptides.
Given the growing number of PPIs involved in human diseases, it has become evident that expanding the toolbox of chemical probes is needed to accurately dissect the precise mechanisms in which PPIs contribute to disease. Herein, we have developed a new and facile design to macrocyclize and staple peptide probes using our FTDR platform that offers high chemoselectivity and enhanced cell permeability compared to traditional approaches.
More details about this work can be found here: “Unprotected peptide macrocyclization and stapling via a fluorine-thiol displacement reaction” in Nature Communications, https://www.nature.com/articles/s41467-022-27995-5
References
- Ryan, D. P. & Matthews, J. M. Protein–protein interactions in human disease. Curr. Opin. Struct. Biol. 15, 441 –446 (2005).
- Lu, H., Zhou, Q., He, J. et al. Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Sig Transduct Target Ther. 5, 213 (2020).
- Moiola, M., Memeo, M. G., & Quadrelli, P. Stapled Peptides-A Useful Improvement for Peptide-Based Drugs. Molecules. 24, 3654 (2019).
- Loren D. Walensky & Gregory H. Bird. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem. 57, 6275–6288 (2014).
- Wang et al. Steric-free bioorthogonal labeling of acetylation substrates based on a fluorine-thiol displacement reaction (FTDR). J. Am. Chem. Soc. 143, 1341-1347 (2021).
- Kobayashi, T., Hoppmann, C., Yang, B. & Wang, L. Using protein-con fined proximity to determine chemical reactivity. J. Am. Chem. Soc. 138, 14832 –14835 (2016).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in