A mutation-induced drug resistance database (MdrDB)
Published in Research Data

By launching this database we hope to facilitate the advancement of the understanding of mutation-induced drug resistance, the development of combination therapies, and the discovery of novel chemicals."
Background
The structural mutation of proteins can directly affect their folding and stability, function, interactions with other proteins and binding affinity. In some cases, it can result in significant perturbations to---or even complete abolishment of---protein function, potentially leading to disease or cancer1. The evolutionary pressure imposed by small molecule drugs on many quickly evolving systems, including cancer cells, viruses, and bacteria, can lead to the rapid development of resistance2. While novel and cheap high-throughput sequencing technologies have made it possible to identify mutations in large populations, the significance and characteristics of any novel polymorphisms currently require time-consuming and expensive experiments to determine3. Protein-ligand binding affinity data is of great value for understanding the impact of polymorphisms on disease and identifying mutations that lead to drug resistance4. Convenient and broad access to such data for wild type and mutant proteins would aid our understanding of the mechanisms of mutation-induced drug resistance, increase the accuracy of extrapolations to novel mutations and systems, and enable more effective computational approaches for drug resistance prediction.
About MdrDB
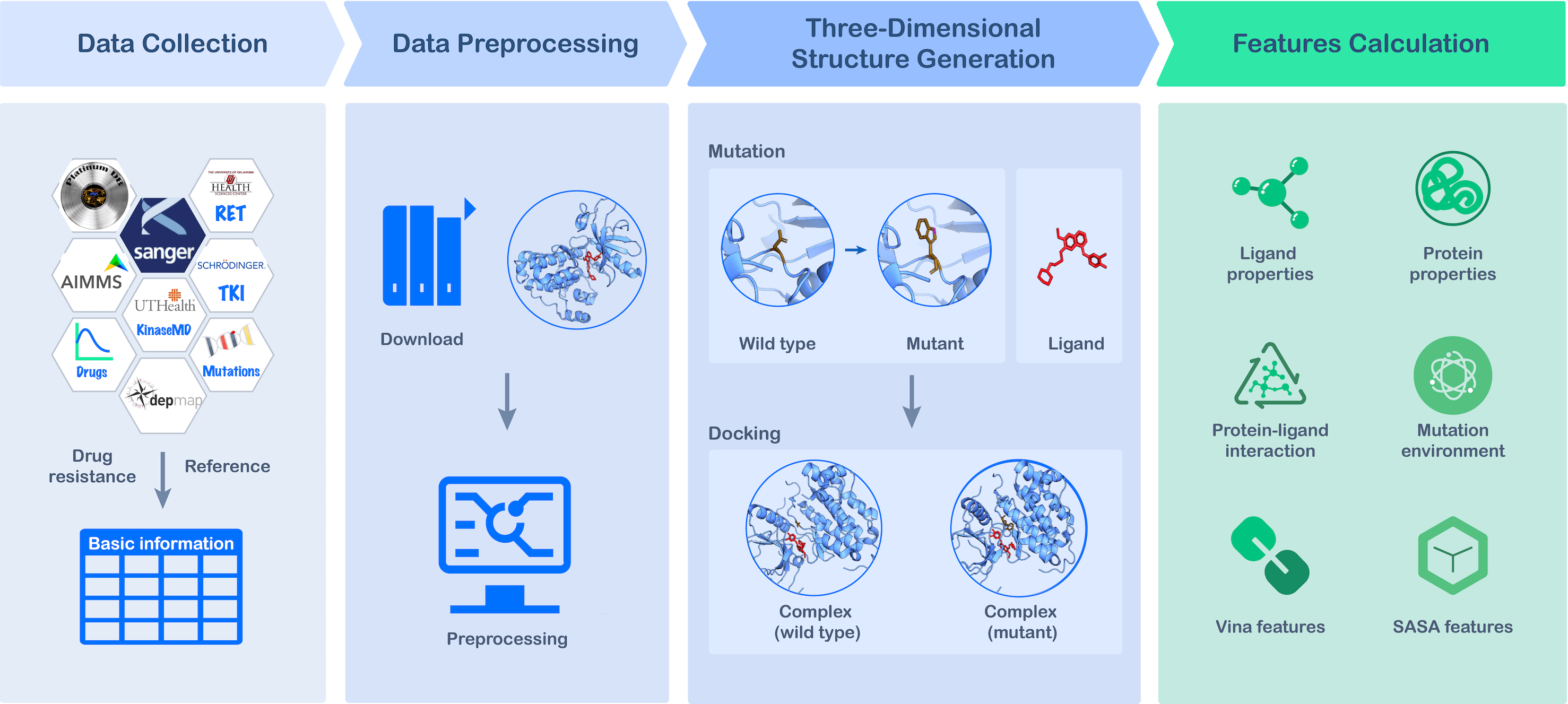
In our latest work published in Communications Chemistry, we created MdrDB, a comprehensive database that significantly expands on the amount of information on drug resistance available to researchers. It brings together wild type protein-ligand complexes, mutant protein-ligand complexes, binding affinity changes upon mutation (ΔΔG), and biochemical features calculated from complexes to advance our understanding of mutation-induced drug resistance, the development of combination therapies, and the discovery of novel chemicals.
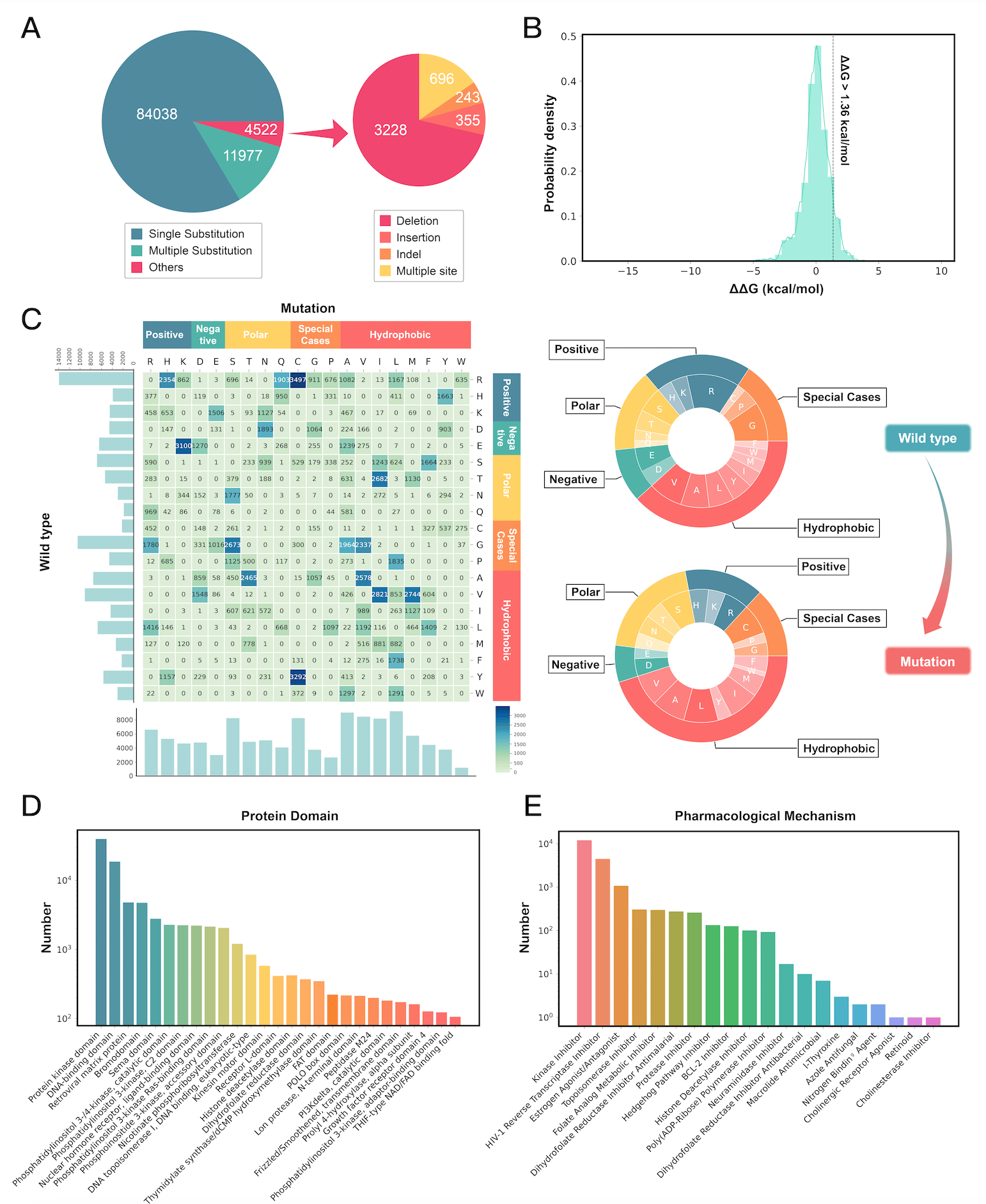
At the time of writing, MdrDB contains 100,537 samples, generated from 240 proteins (5,119 total PDB structures), 2,503 mutations, and 440 drugs. In addition to single-point substitution mutations and multiple-points substitution mutations, MdrDB also contains complex mutations including deletion, insertion, and indel (insertion-deletion) mutations, as well as multiple-site mutations containing a number of the aforementioned mutations.
We have developed a user-friendly website, https://quantum.tencent.com/mdrdb/, to provide access to the curated data and structural information on wild type and mutant complexes, and allow users to browse, search, display and download the data.
Advantages
MdrDB offers several key advantages over existing publicly available protein mutation databases:
● A comprehensive database: MdrDB is the largest mutation-induced drug resistance database and integrates information from multiple sources, covering mutations across various protein families. A database of this size and breadth enables the development of more effective data-driven models for drug resistance prediction.
● Structure-based: MdrDB provides 3D structural information on all the wild type and mutant proteins included in the database. Such structural information is critical for both accurate feature calculations used in computational drug design as well as the visualization of the effects of mutation. Based on these structures, a total of 146 biochemical features were calculated to provide data for drug resistance modeling.
● Diverse protein mutations: In addition to single-point and multi-site substitution mutations considered in existing databases, MdrDB contains a variety of complex mutation types such as deletions, insertions, and insertion-deletions, which can play an important role in some disease progressions.
Drug resistance modeling
In addition to the development of the database itself, in this work, we also evaluated the impact of using MdrDB and other publicly available drug resistant databases as training data on the performance of 10 classical machine learning models in predicting tyrosine kinase inhibitors affinity change values. By using MdrDB as the training set, we found that nearly all models gained significant performance improvement. Furthermore, we also performed a comprehensive evaluation of those 10 models in several different scenarios, and provide baseline prediction results on the MdrDB database. It is our hope that these benchmarking studies will help further the development of new machine learning algorithms using the MdrDB database and facilitate drug resistance research.
We created MdrDB specifically to have the size, breadth, and complexity to be useful for practical protein mutation studies and drug resistance modeling and, as more public data becomes available in the future, MdrDB will continue to be updated regularly.
● Our full paper is available at: https://www.nature.com/articles/s42004-023-00920-7.
● All data is available to browse and download on the MdrDB website: https://quantum.tencent.com/mdrdb/.
● A full tutorial for MdrDB is available at: https://quantum.tencent.com/mdrdb/tutorial.
References
1. Li, M., Petukh, M., Alexov, E. & Panchenko, A. R. Predicting the impact of missense mutations on protein–protein binding affinity. J. chemical theory computation 10, 1770–1780 (2014).
2. Cohen, M. L. Epidemiology of drug resistance: implications for a post—antimicrobial era. Science 257, 1050–1055 (1992).
3. Consortium, I. C. G. et al. International network of cancer genome projects. Nature 464, 993 (2010). 16. MacLean, D., Jones, J. D. & Studholme, D. J. Application of’next-generation’sequencing technologies to microbial genetics. Nat. Rev. Microbiol. 7, 96–97 (2009).
4. Hauser, K. et al. Predicting resistance of clinical abl mutations to targeted kinase inhibitors using alchemical free-energy calculations. Commun. biology 1, 1–14 (2018).
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in