A recyclable stereoauxiliary aminocatalyzed strategy for one-pot synthesis of indolizine-2-carbaldehydes
Published in Chemistry
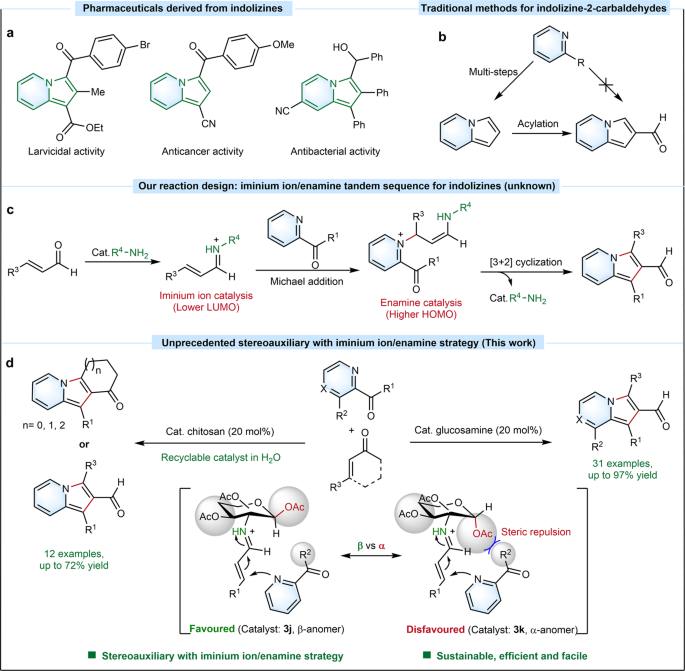
Indolizines are a crucial group of N-heterocyclic compounds that have a wide range of applications in various fields.1-3 The aldehyde group in the pyrrole ring makes indolizine-2-carbaldehydes versatile building blocks, but one-pot synthesis and synthetic modifications of these compounds have not been well-studied due to the lack of efficient strategies.4-5 A one-pot synthesis method for indolizine-2-carbaldehydes was first reported in 2021 using an acetic acid-catalyzed approach.6 However, a more generalized and efficient synthesis strategy is still desired to broaden the scope of indolizine-2-carbaldehydes with improved efficiency via [3+2] cyclization.
Aminocatalysis via iminium ion or enamine is a significant approach for constructing C-C bonds that has been developed in recent decades.7-8 We propose iminium ion/enamine tandem sequence that could efficiently overcome energy barriers encountered in Michael and aldol reactions during [3+2] annulations of acyl pyridines and α,β-unsaturated aldehyde for the construction of indolizine-2-carbaldehydes (Fig. 1a). Furthermore, inspired by our recently work on anomeric stereoauxiliary cleavage of the C−N bond of glucosamine for the efficient preparation of imidazo[1,5-a]pyridines,9 D-glucosamine and polymeric chitosan derived from biomass containing mostly β-D-anhydroglucosamine units are used as stereoauxiliary catalysts to enhance the efficiency of the [3+2] annulations (Fig. 1b).
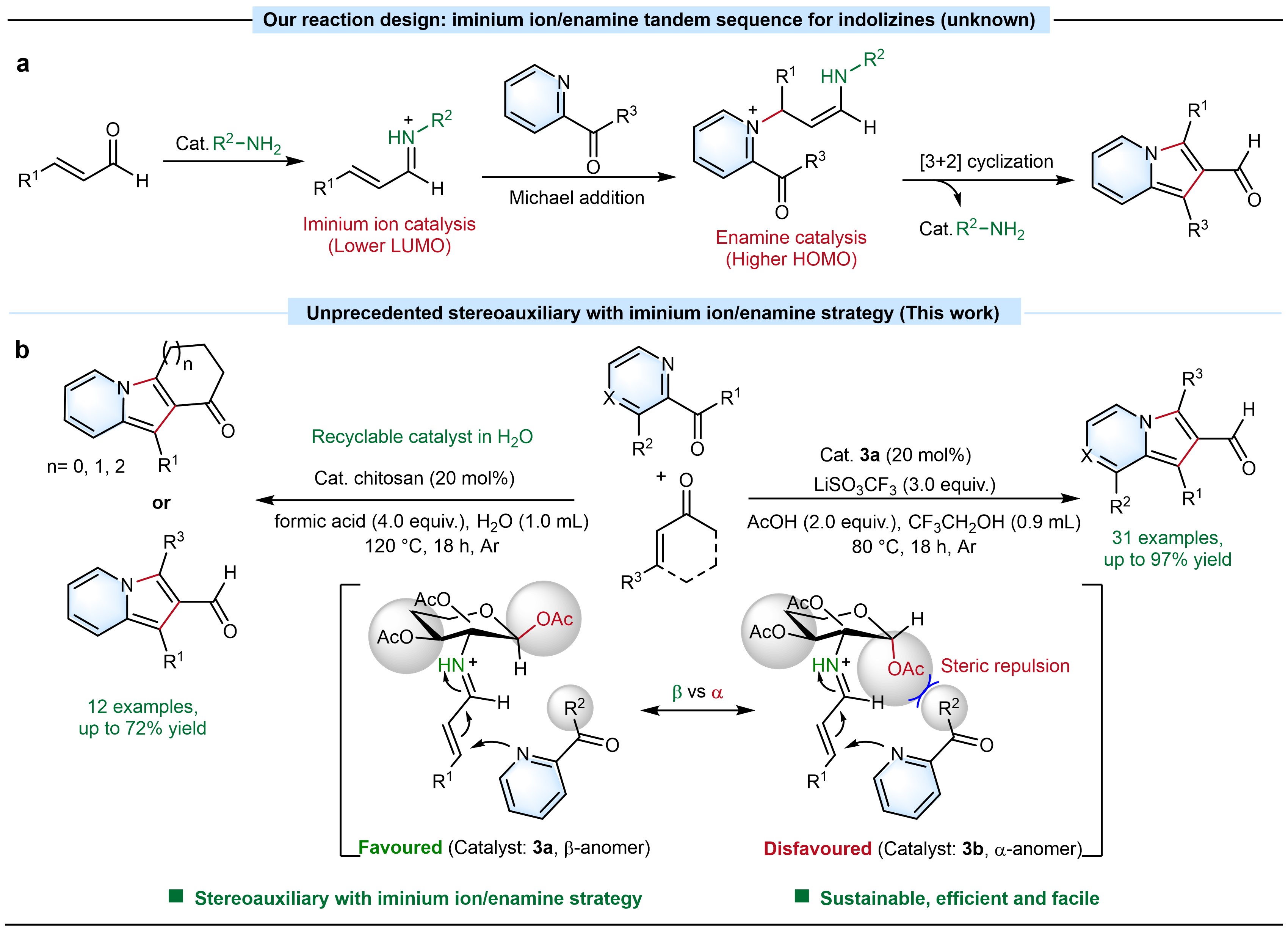 Fig. 1 a) Our design: iminium ion/enamine tandem sequence strategy for trisubstituted indolizine-2-carbaldehydes. b) This work: Unprecedented stereoauxiliary aminocatalysis with iminium ion/enamine strategy for the preparation of 1,2,3-trisubstituted indolizine-2-carbaldehydes via one-pot reaction.
Fig. 1 a) Our design: iminium ion/enamine tandem sequence strategy for trisubstituted indolizine-2-carbaldehydes. b) This work: Unprecedented stereoauxiliary aminocatalysis with iminium ion/enamine strategy for the preparation of 1,2,3-trisubstituted indolizine-2-carbaldehydes via one-pot reaction.
To explore the mechanism, under standard conditions, catalyst 3a with β-anomer had a higher yield (97%) of the desired product 4 compared to catalyst 3b with α-anomer (53%). Furthermore, imine intermediates were synthesized and tested to gain insight into the reaction mechanism, which showed that the β-anomer (3c) achieved a higher yield (51%) compared to the α-anomer (3d) with only 16% yield of 4. This verifies that the reaction pathway via aminocatalyst preferentially reacts with α,β-unsaturated aldehydes and that the stereoauxiliary effect from the β-anomer promotes the yield of 4. Additionally, the lower yield with the α-anomer (3d) supports the existence of steric hindrance from the acetyl group at the C1 position. The results of these control experiments verified the stereoauxiliary effect favored by the β-anomer and the steric shielding effect from the α-anomer.
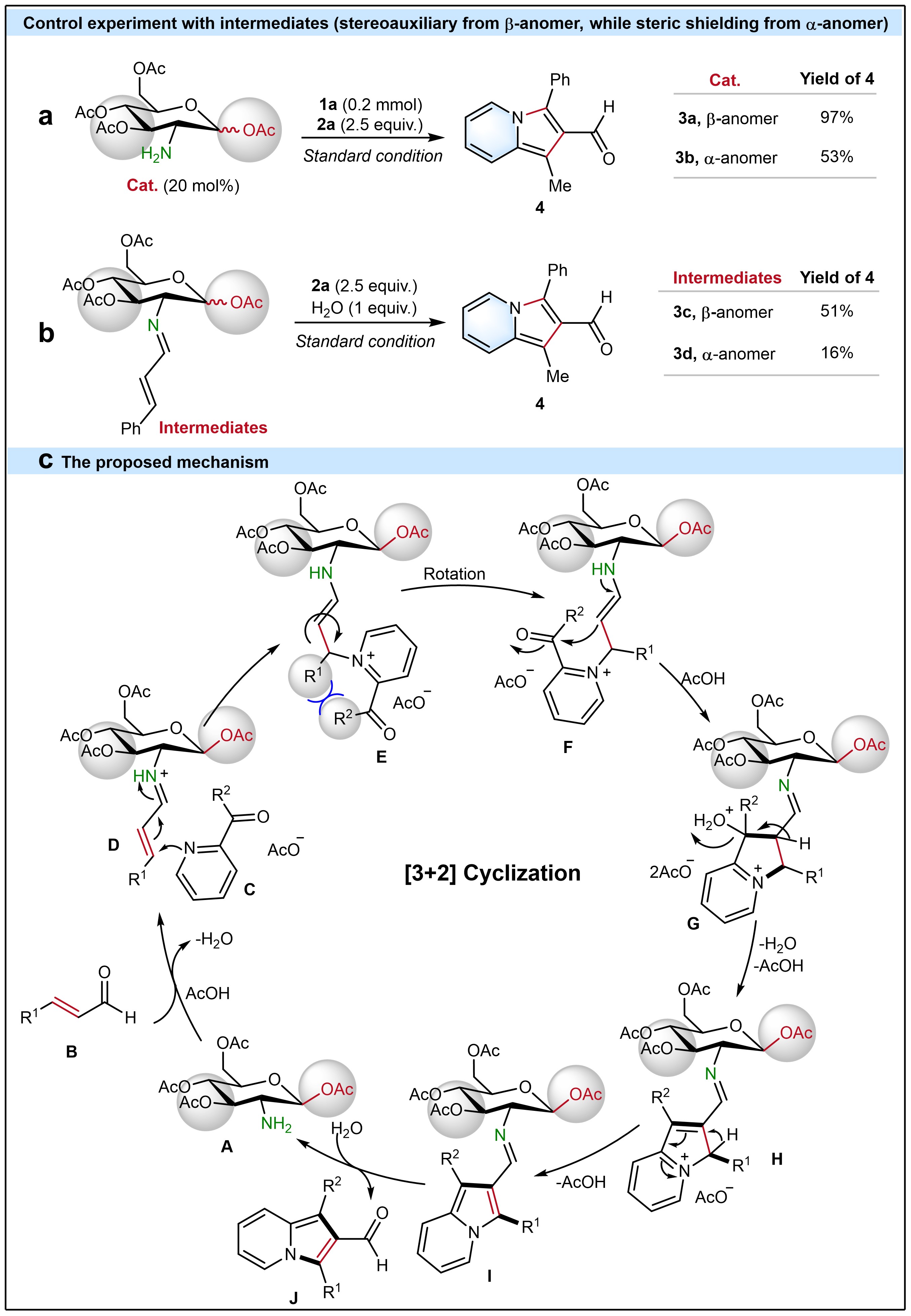 Fig. 2 Stereoauxiliary control experiments. a) Control experiment with 3a (β-anomer) and 3b (α-anomer). b) Control experiment with intermediate 3c (β-anomer) and intermediate 3d (α -anomer). c) Proposed mechanism.
Fig. 2 Stereoauxiliary control experiments. a) Control experiment with 3a (β-anomer) and 3b (α-anomer). b) Control experiment with intermediate 3c (β-anomer) and intermediate 3d (α -anomer). c) Proposed mechanism.
Combining all results, a plausible mechanism is proposed (Fig. 2c). First, aminocatalyst A reacts with α,β-unsaturated aldehyde B to form iminium ion D.7 Then, 2-acetylpyridine attacks the iminium ion D via Michael addition reaction to generate an enamine E.7,10 Enamine F can be simply converted from E via the rotation, which will overcome the bulky steric hindrance between R1 and R2. Thereafter, an intermediate G forms via the intramolecular cyclization reaction in the enamine F. Then, an intermediate H generates from the intermediate G through a dehydration reaction, which leads to an intermediate I after deprotonation. Finally, the desired indolizine-2-aldehyde J forms via the hydrolysis reaction of intermedidate I and the catalyst A is regenerated (ESI-HRMS: m/z calcd. for C14H22NO9+ [M]: 348.1289, found 348.1297. The conformation stability of catalyst 3a was proved with 1H NMR) for the next catalysis cycle. Computational investigations of the mechanistic and stereochemical aspects of this study are underway in the Houk lab at UCLA.
The study explored the scope of the aminocatalyzed reaction using various α,β-unsaturated aldehydes and heteroaryl ketones substrates, including electron-donating or -withdrawing groups, and aliphatic, aromatic, and heteroaryl substrates. The reaction was well-tolerated with various valuable functional groups at different positions of the substrates and delivered high yields of the desired products. The structure of one product was confirmed by X-ray crystallographic analysis. To test the applicability of indolizine-2-carbaldehydes for the preparation of indolizine with important biological activities, a group of important bioactive molecules or drugs was used under our protocol. For example, an important fluvastatin intermediate was tolerant with the standard condition for the preparation of indolizine-2-carbaldehyde (5). (E)-3-(4-hydroxy-3-methoxyphenyl)acrylaldehyde from Gliricidia sepium was accessed by our method with 79% yield of 3-(4-hydroxy-3-methoxyphenyl)-1-(pyridin-2-yl)indolizine-2-carbaldehyde (6). Interestingly, (E)-3-(4-(dimethylamino)phenyl) acrylaldehyde that is often used to detect catechins11 was also smoothly transformed into 3-(4-(dimethylamino)phenyl)-1-(pyridin-2-yl)indolizine-2-carbaldehyde (7, 81%). Furthermore, the obtained indolizine-2-carbaldehydes can be easily modified in later stages to provide more complex molecules efficiently (Fig. 3b). For example, 3-(4-bromophenyl)-1-(pyridin-4-yl)indolizine-2-carbaldehyde (8) underwent successful reduction (9), arylation (10), condensation (11) or dehydration [5+1] annulations (12), to showcase the synthetic diversifications on 1,2,3-trisubstituted indolizine-2-carbaldehydes.
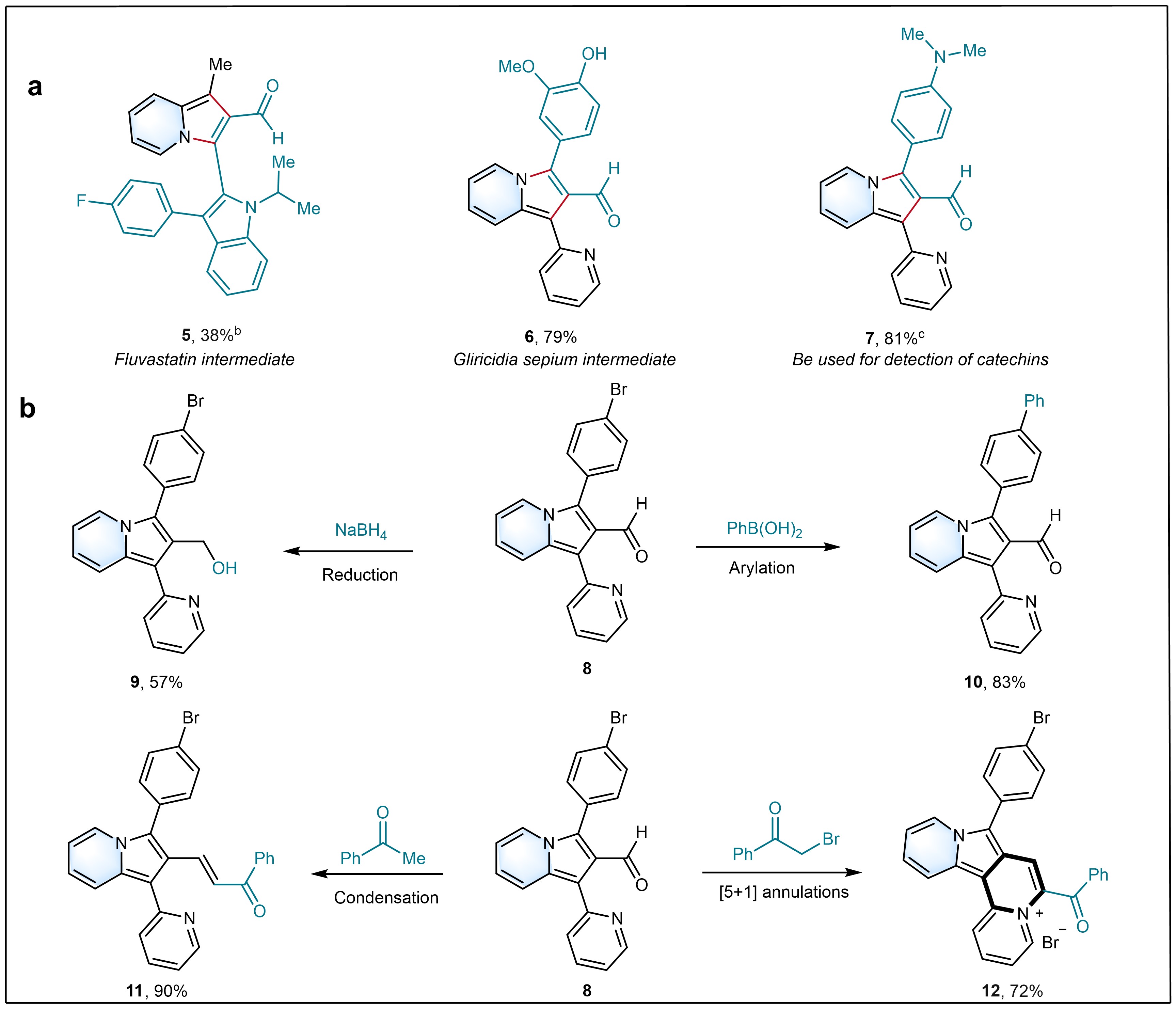 Fig. 3 Synthetic applications. a) Late-stage selective modifications of bioactive molecules and drugs. b) Late-stage diversification. aYields are those of isolated products. bReaction for 42 h in AcOH : CF3CH2OH (0.45 : 0.45 mL). cReaction for 42 h in AcOH : CF3CH2OH (0.4 : 0.5 mL).
Fig. 3 Synthetic applications. a) Late-stage selective modifications of bioactive molecules and drugs. b) Late-stage diversification. aYields are those of isolated products. bReaction for 42 h in AcOH : CF3CH2OH (0.45 : 0.45 mL). cReaction for 42 h in AcOH : CF3CH2OH (0.4 : 0.5 mL).
A recyclable aminocatalyst was used to obtain various indolizine-aldehyde products in an efficient and sustainable manner using the biopolymer chitosan, which is made of β-D-glucosamine building blocks. The use of chitosan demonstrated a recyclable aminocatalysis strategy and the reaction was highly efficient in H2O without the need for lithium salts. Various indolizine-aldehydes and cyclic α,β-unsaturated ketones were obtained using chitosan as aminocatalyst, with higher yields for some products compared to using glucosamine. Our anomeric stereoauxiliary aminocatalyst was compared with a state-of-the-art method6 and found to be more robust with higher efficiency in H2O and high yields for various indolizine-aldehydes, even for sensitive groups. Our protocol was also successfully applied on a larger scale and chitosan showed excellent catalytic performance even after 3 catalytic cycles. The product can be easily isolated and the remaining aqueous phase can be reused for the next catalytic cycle.
References
- Majumdar, K. C. & Chattopadhyay, S. K. Heterocycles in Natural Product Synthesis. (John Wiley & Sons, 2011).
- Kim, E., Lee, Y., Lee, S. & Park, S. B. Discovery, understanding, and bioapplication of organic fluorophore: a case study with an indolizine-based novel fluorophore, Seoul-Fluor. Chem. Res. 48, 538-547 (2015).
- Sharma, V. & Kumar, V. Indolizine: a biologically active moiety. Chem. Res. 23, 3593-3606 (2014).
- Biagetti, M., Accetta, A., Capelli, A. M., Guala, M. & Retini, M. Indolizine derivatives as phosphoinositide 3-kinases inhibitors. US patent No. US 2015/0361100 A1. (17-12-2015).
- Burnett, D., A.; Vacca, Joseph, P. Compounds, compositions and methods of use. International patent No. WO 2018/195075 A1. (25-10-2018).
- Yuan, Y.-C., Liu, T.-Z. & Zhao, B.-X. Metal-Free Catalyzed Synthesis of Fluorescent Indolizine Derivatives. Org. Chem. 86, 12737-12744 (2021).
- Erkkilä, A., Majander, I. & Pihko, P. M. Iminium catalysis. Rev. 107, 5416-5470 (2007).
- Chauhan, P., Mahajan, S. & Enders, D. Achieving molecular complexity via stereoselective multiple domino reactions promoted by a secondary amine organocatalyst. Chem. Res. 50, 2809-2821 (2017).
- Zeng, K., Ye, J., Meng, X., Dechert, S., Simon, M., Gong, S., Mata, R.A. & Zhang, K. Anomeric stereoauxiliary cleavage of the C‐N bond of D‐glucosamine for the preparation of imidazo [1, 5‐α] pyridines. Eur. J. 28, e202200648 (2022).
- Moyano, A. & Rios, R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Rev. 111, 4703-4832 (2011).
- Glavnik, V., Simonovska, B. & Vovk, I. Densitometric determination of (+)-catechin and (−)-epicatechin by 4-dimethylaminocinnamaldehyde reagent. Chromatogr. A 1216, 4485-4491 (2009).
Follow the Topic
-
Communications Chemistry

An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.
Related Collections
With Collections, you can get published faster and increase your visibility.
f-block chemistry
Publishing Model: Open Access
Deadline: Feb 28, 2026
Experimental and computational methodology in structural biology
Publishing Model: Open Access
Deadline: Apr 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in