A Trifluoromethyl- and Fluoro-Substituted Stereogenic Carbon Centre Gives Rise to Unorthodox Chemistry
Published in Chemistry

Fluorine, although the most abundant halogen in earth’s crust, is scarcely found in a natural product1. This makes laboratory synthesis the most viable way of accessing fluoro-organic compounds2, which are crucial to drug discovery, advances in agrochemicals, and materials research. Among other attributes, a fluoro substituent can enhance membrane permeability and metabolic stability3. The three pairs of electrons of a densely-packed fluorine atom can engender strong electrostatic interactions, and the lower energy σ*C–F orbitals encourage hyperconjugation. Molecules bearing a trifluoromethyl- or a fluoro-substituted stereogenic center are thus a mainstay in therapeutic science. In contrast, little is known about compounds containing a carbon connected to a trifluoromethyl and a fluorine atom. We reasoned that a catalytic method for synthesising homoallylic alcohols with a polyfluoro-substituted stereogenic carbon centre would make it possible to access and evaluate new analogues of several important drugs (Fig. 1), including Gemzar (cancer chemotherapy)4, Sovaldi (chronic hepatitis C virus infection)5,6,7,8,9, Clolar (anti-leukemic)10, or bioactive entities such as sialyltransferase inhibitor11,12 3Fax-Neu5Ac.
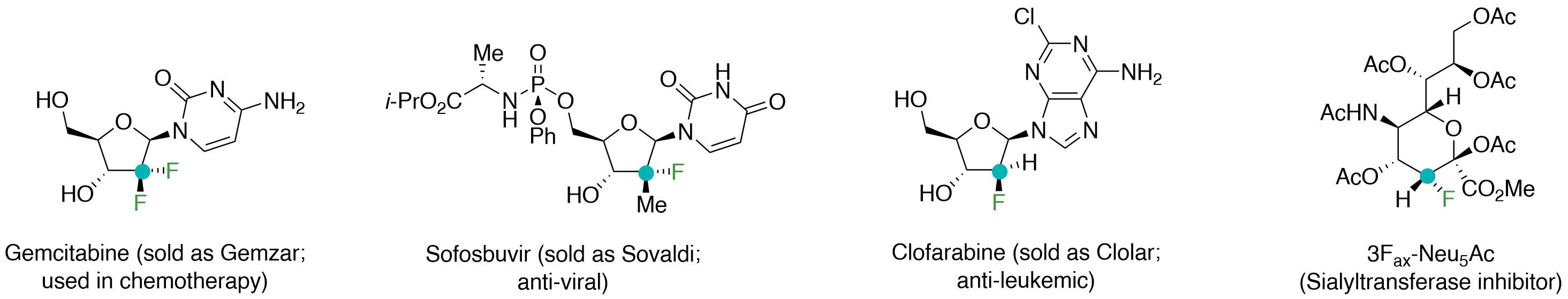
Figure 1. Representative bioactive organofluorine compounds, variants of which, bearing a trifluoromethyl- and fluoro-substituted carbon, would be intriguing leads for drug discovery.
We opted for an amino acid-based catalyst system13, formerly used for α- and enantioselective additions of allylboronates to aldehydes14, with polyfluoro allylboronate 1 as the nucleophile precursor (Fig. 2). The expectation was that 2 (need α-addition) would be favoured, requiring us somehow to coax the process to be not only -selective, but deliver what we believed to be the less preferred S,R-3, the isomer needed for polyfluoro monosaccharide synthesis. Startlingly, S,R-3 was formed directly, the same holding for other aldehydes (Fig. 2). It took several x-ray structures to convince us about the outcome. Our mechanistic hypotheses needed an overhaul, but we felt lucky.
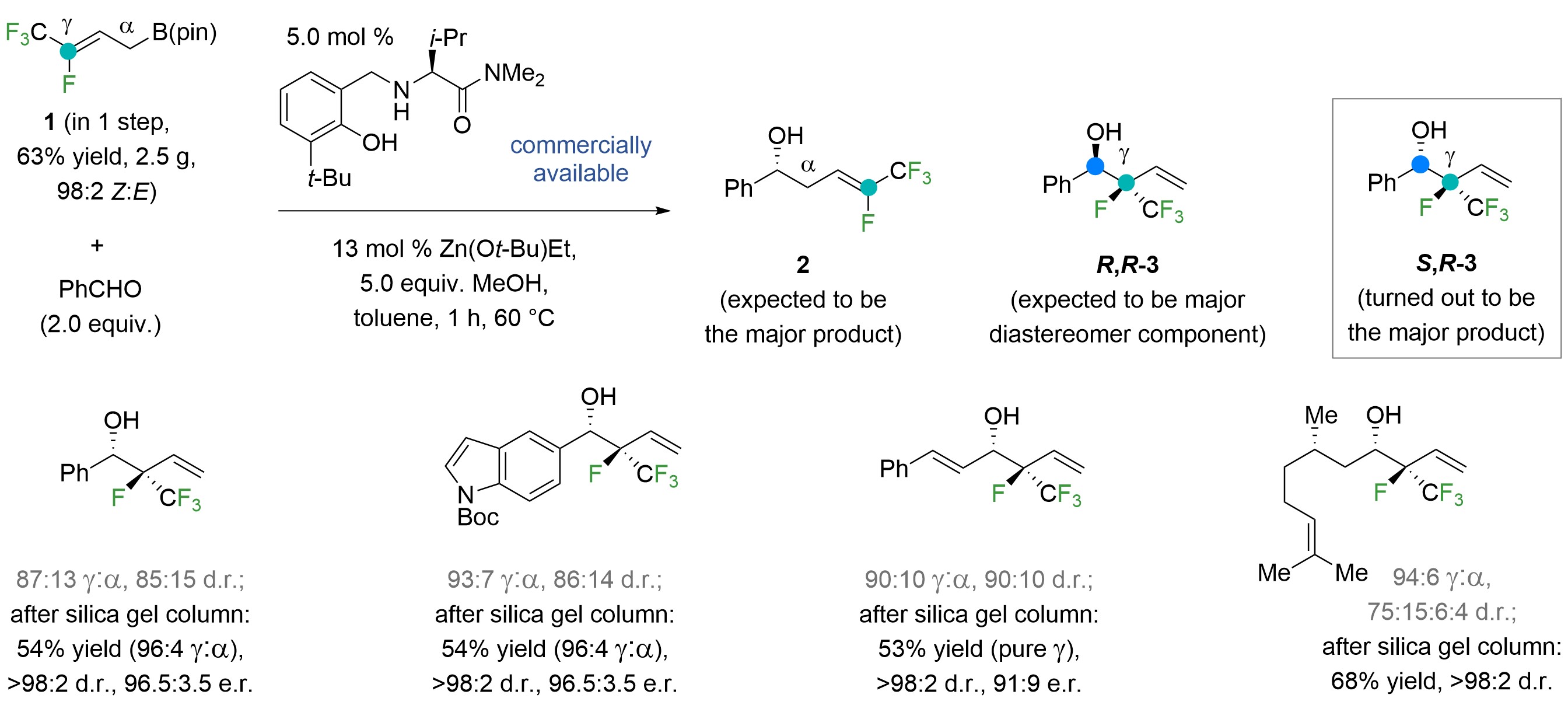
Figure 2. The catalytic method had two (auspicious) surprises: the addition is γ-selective and affords the diastereomer needed for applications to polyfluoro monosaccharides.
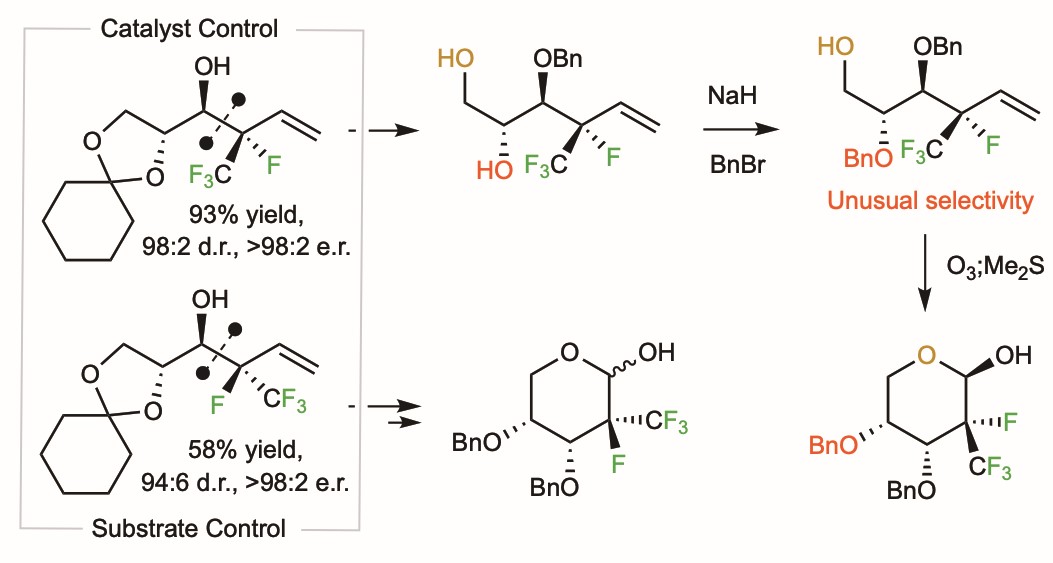
To see if the catalytic process can provide access to polyfluoro furanoses and hexoses (Fig. 3), we reacted 1 with aldehyde 4, prepared the benzyl ether, and removed the acetal, obtaining 5. Hardly expecting another shocker, we took what we thought would be the first step in a graceless protection/deprotection sequence leading to differentially protected 6, which would then be converted to pyran 7. However, it was 6 that was formed when 5 was subjected to NaH and BnBr (54% yield): the more hindered alcohol had indeed reacted preferentially. Analogously, reaction of diast-5, obtained from a substrate-controlled stereoselective process involving 1 and 415, afforded diast-6 as the major isomer (39% yield). We were happy to be able to access different polyfluoro heterocycles efficiently. In the meantime, though, another brain teaser had landed on our lap.
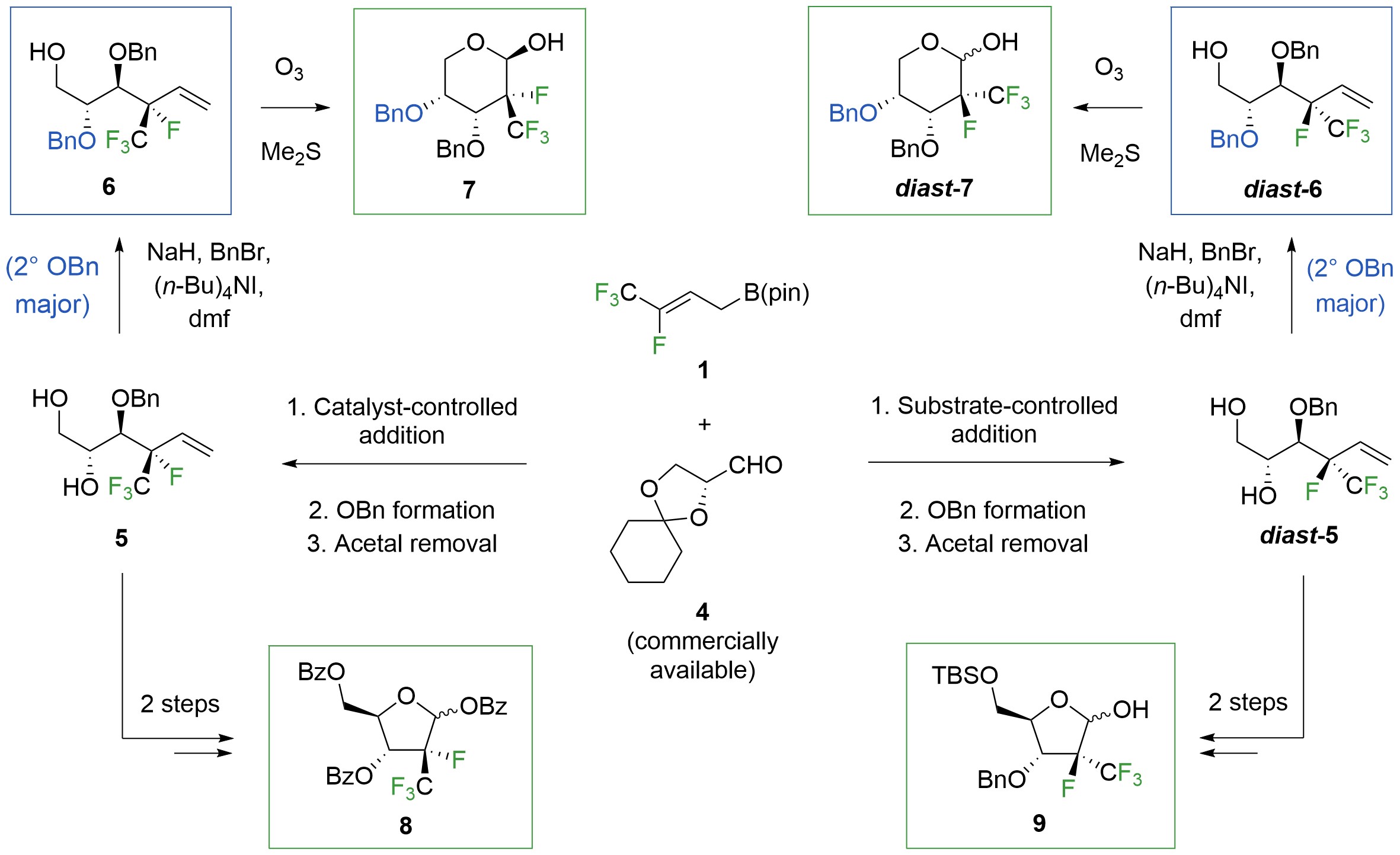
Figure 3. Synthesis of polyfluoro monosaccharides proved to be more efficient than expected, owing to yet another surprise discovery.
To clarify the mechanistic riddles, we carried out an assortment of experiments and benefitted from the computational expertise of Professor Liu and his team at Pittsburgh. Before long, it was evident that the catalyst is not an organoboron species, as we had supposed, but probably an organozinc compound (Fig. 4). The longer distances in a Zn complex alleviate steric and electronic repulsions and 1,3-shift transition state becomes easier, especially with fluorine atoms around. The initially formed Zn-allyl-3° can react directly with an aldehyde (via ts-α-addn; Fig. 4), giving the -addition isomer (initially expected), but the fluorine atoms probably destabilize ts-α-addn. This accelerates rearrangement to the less congested Zn-allyl-1°, and reaction via ts-γ-addn-1 becomes the ensuing favored option (vs. ts-γ-addn-2). Control experiments show that even with only the trifluoromethyl group present (i.e., H instead of F), the process reverts to being α-selective.
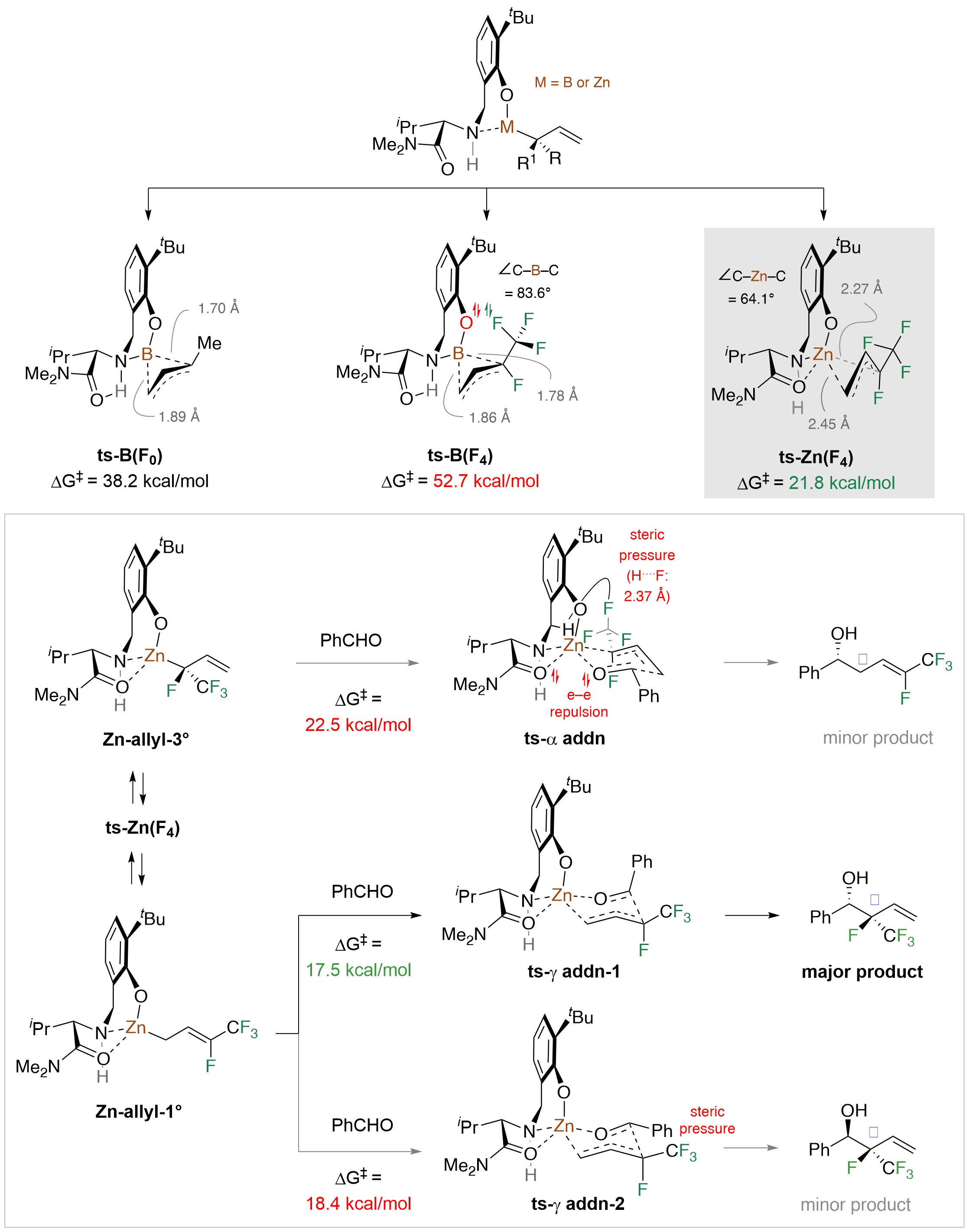
Figure 4. Computational studies indicate that the catalyst is likely an organozinc – not an organoboron – compound, and offer explanations regarding the unexpected regio- and stereoselectivity profiles of the catalytic process.
The uncommon preference for reaction at the more congested secondary alcohols also originates from the polyfluoro site. In ts-sec-1 (Fig. 5), electronic repulsion raises the latter’s nucleophilicity (higher ground state). For the other diol isomer, reaction preferentially via ts-sec-2’ is favored: the key interaction being H-bonding between the oxy-anion and the alkenyl H, rendered acidic by its fluorine neighbours (lower transition state). Here too, without having both a trifluoromethyl and a fluorine substituent, it is the primary alcohol that is preferentially protected.
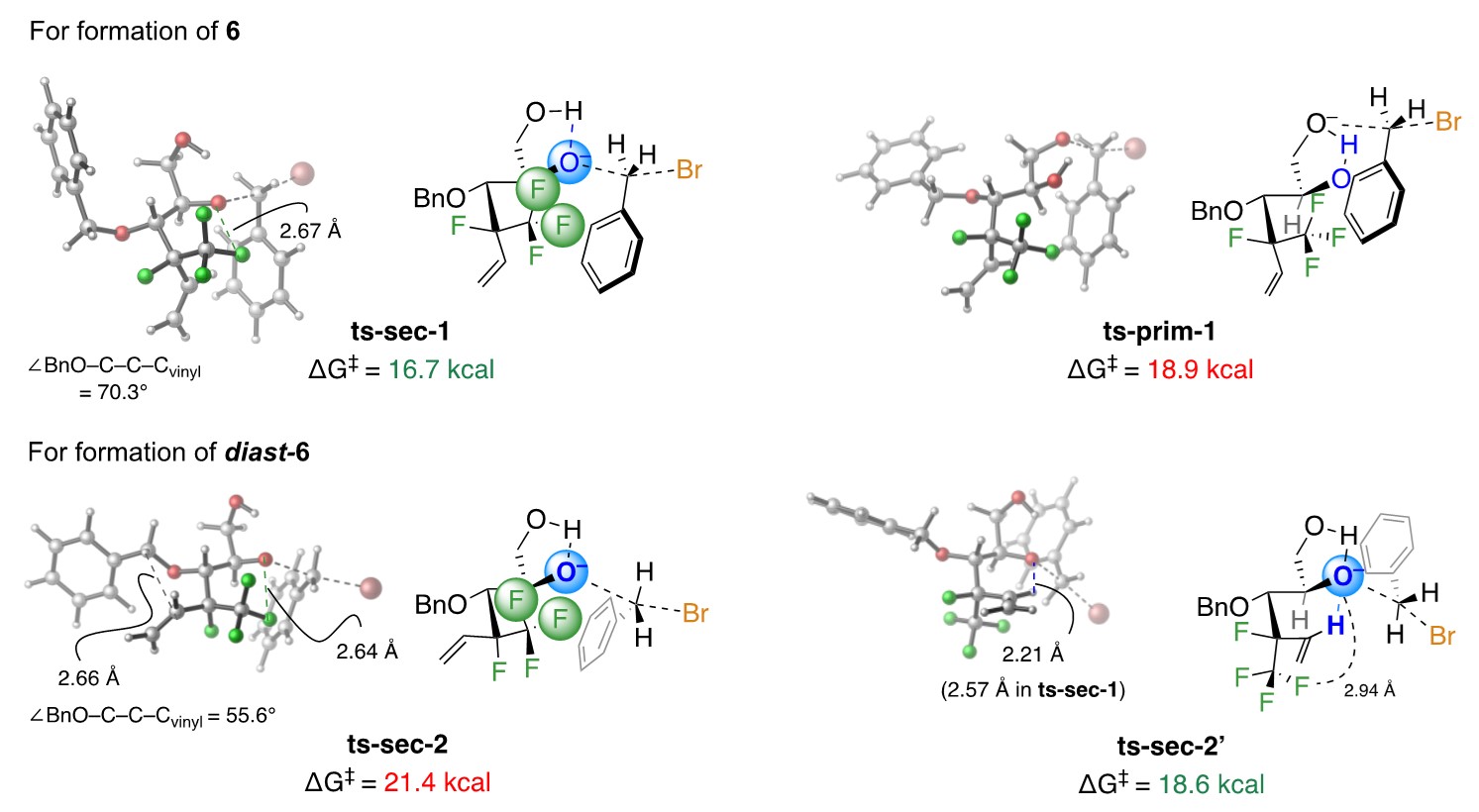
Figure 5. Experimental and computational studies offer an answer to a puzzle: why does a secondary alcohol adjacent to a polyfluoro site react faster than a primary alcohol?
What spurred us was the hope of finding a practical and selective way of synthesising homoallylic alcohols with a trifluoromethyl- and fluoro-substituted allylic carbon, and by doing so, help accelerate drug discovery. The idea was that four C–F bonds and 12 pairs of nonbonding electrons in a confined space can cause unusual, and hopefully beneficial, molecular interactions. We should have anticipated that the same would hold for the intrinsic chemistry of these fascinating and poorly understood molecules.
Contributors
Author
Amir H. Hoveyda
Professor of Chemistry and Director of Laboratories for Catalytic Chemical Synthesis, Boston College and University of Strasbourg
Contributing authors
Shibo Xu
Graduate Student, Boston College
Juan del Pozo
Former postdoc, Boston College (presently at Merck, Sharp and Dohme)
References:
- O’Hagan, D. et al. Biosynthesis of an organofluorine molecule. Nature 416, 279–279 (2002).
- Paioti, P. H. S. et al. Catalytic and stereoselective transformations with easily accessible and purchasable allyl and alkenyl fluorides. Angew. Chem. Int. Ed. doi:10.1002/anie.202208742.
- Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
- Hertel, L. W. et al. Evaluation of the antitumor activity of gemcitabine (2’,2’-difluoro-2’-deoxycytidine). Cancer Res. 50, 4417–4422 (1990).
- Sofia, M. J. et al. Discovery of a b-D-2’-deoxy-2’-a-fluoro-2’-b-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53, 7202–7218 (2010).
- Fung, A. et al. Efficiency of incorporation and chain termination determines the inhibition potency of 2’-modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob. Agents and Chemother. 58, 3636–3645 (2014).
- Keating, G. M. Sofosbuvir: a review of its use in patients with chronic hepatitis C. Drugs. 74, 1127–1146 (2014).
- Appleby, T. C., et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 347, 771–775 (2015).
- Mengshetti, S. et al. Discovery of a series of 2’-a-fluoro,2’-b-bromo-ribonucleosides and their phosphoramidate prodrugs as potent pan-genotypic inhibitors of hepatitis C virus. J. Med. Chem. 62, 1859–1874 (2019).
- Bonate, P. L. et al. Discovery and development of clofarabine: a nucleoside analogue for treating cancer. Nat. Rev. 5, 855–863 (2006).
- Rilahan, C. D. et al. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Bio. 8, 661–668 (2012).
- Büll, C. et al. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano 9, 733–745 (2015).
- Silverio, D. L. et al. Simple organic molecules as catalysts for enantioselective synthesis of amines and alcohols. Nature 494, 216–221 (2013).
- Morrison, R. J., van der Mei, F. W., Romiti, F. & Hoveyda, A. H. A catalytic approach for enantioselective synthesis of homoallylic alcohols bearing a Z-alkenyl chloride or trifluoromethyl group. A concise and protecting group-free synthesis of mycothiazole. J. Am. Chem. Soc. 142, 436–447 (2020).
- Chen, J. L.-Y., et al. Highly diastereo- and enantioselective allylboration of aldehydes using a-substituted allyl/crotyl pinacol boronic esters via in situ generated borinic esters. J. Am. Chem. Soc. 135, 5316–5319 (2013).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.






Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in