Accelerating and improving drug discovery by transfer learning in the multi-fidelity setting
Published in Chemistry, Protocols & Methods, and Computational Sciences
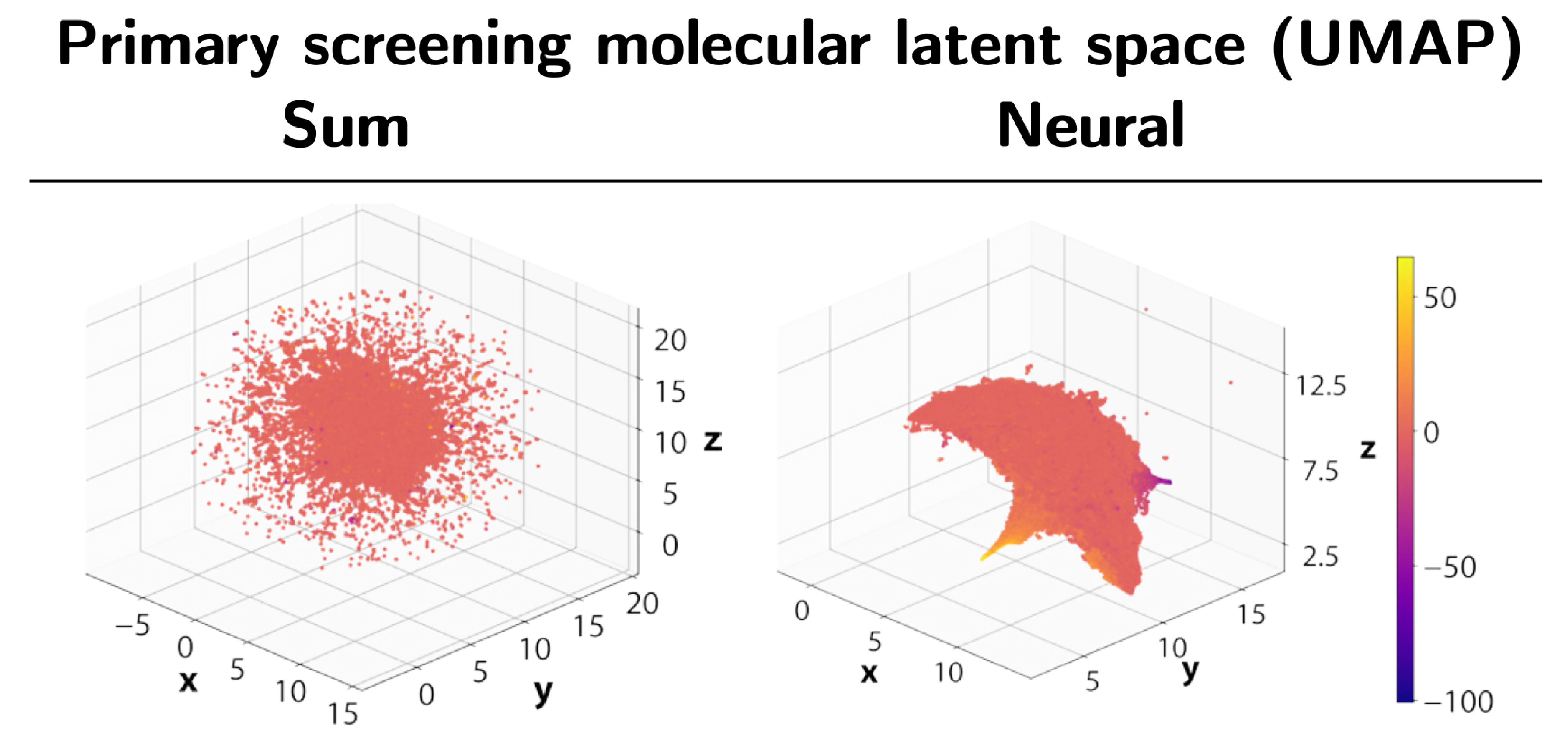
Modern molecular discovery processes can generate millions of measurements in a very short amount of time. However, not all measurements are equally useful. To balance resource utilisation, costs, and accuracy, most workflows are designed as a funnel or cascade of operations at different fidelity levels. Millions of cheap but approximate measurements are performed as a starting point, which are then followed by increasingly more accurate and expensive experiments.
The nature and requirements of molecular discovery can be vastly different depending on the specific application. However, the funnel design has been widely adopted by practitioners as it can cheaply and effectively guide the more expensive experiments towards regions of interest. This strategy can be applied both to wet lab and computational problems. A prominent example of the first is drug discovery by high-throughput screening (HTS). At a high-level, HTS experimentally determines the activity of molecules against a target of therapeutic interest such as a protein. The molecules are selected from a compound library that is designed to be diverse, and usually contains up to 2 million compounds. However, this first stage of HTS, often referred to as 'primary screening', is limited to measurements made at a single concentration, and is inherently noisy and prone to errors. The process relies heavily on automation, which despite lowering overall costs, incurs certain planning and budget limitations due to its high complexity. Primary screening results are used as a filter for more expensive and accurate screens, usually called 'confirmatory screens'. Such experiments are a richer source of information as they involve measurements at multiple concentrations. At the same time, they are less automated and can only be performed on a small scale. Ultimately, practitioners are interested in the results of confirmatory screens, yielding molecules that can be optimised.

Alternatively, computational pipelines can also adopt a funnel design. This is often encountered for quantum mechanical simulations. Generally, highly accurate computational methods such as at the CCSDT (coupled cluster single-double-triple) level of theory scale poorly with molecule size and are highly prohibitive in terms of computational resource utilisation, limiting experiments to only hundreds of molecules even for most small organic compounds. Higher throughout methods like Density Functional Theory (DFT) can be deployed for hundreds of thousands of molecules; however, this is still feasible only on large computer clusters. Finally, semi-empirical tight-binding models such as GFN2-xTB rely on many simplifications that enable fast calculations even for large systems. It is thus increasingly common to encounter projects that rely not only on one method or level of theory, but a combination of high-throughput and high-precision measurements.
The impact of screening cascades, whether computational or wet lab-based, has consistently been proven over the years. High-throughput screening is one of the key techniques in drug discovery, leading to about a third of clinical candidates and approved drugs. For quantum mechanics, data-driven methods have been at the forefront of machine learning research thanks to their potential to complement or even replace traditional methods. Lately, one of the most promising directions has been integrating calculations from different levels of theory.
Despite the success of traditional drug discovery by HTS, computational methods that support and improve the process have lagged behind. In particular, the millions of measurements performed as part of primary screening are typically discarded after the initial filtering step. To the best of our knowledge, the different tiers or fidelities generated during HTS are not jointly leveraged in any modern machine learning workflow. The use of machine learning for multiple fidelities is more common in the field of quantum mechanics. Compared to drug discovery, where the wet-lab experiments are prone to artefacts and errors, quantum calculations are deterministic and can be described by precise mathematical formulations. For example, one common method in the field is to learn a 'correction term' between measurements at different fidelities, covering the approximate nature of the cheaper methods. However, the application of general purpose deep learning models such as Graph Neural Networks is relatively understudied in the context of multi-fidelity quantum models.
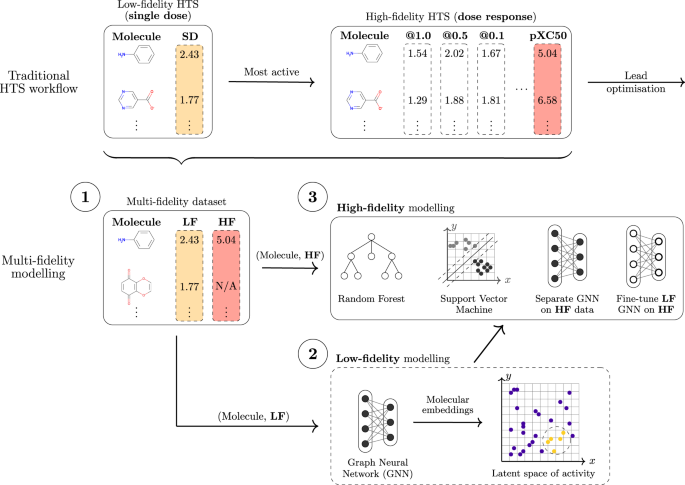
In our work, we tackle the challenge of leveraging the different data types generated during molecular discovery processes. More specifically, for HTS we seek approaches that use millions of primary screening experimentally-derived values to guide models learnt from the more accurate, expensive, and sparse confirmatory-level measurements. Holistically integrating multiple tiers of HTS data has the potential to suggest novel compounds that would otherwise be missed by traditional methods, to lead to a more focused and efficient screening campaign, including time and financial savings, and hybrid wet lab and in silico workflows.
From a machine learning perspective, the problem can be described as a transfer learning task. Metaphorically, we wish to describe a 'chemical universe' based on activities determined during primary screening and use this knowledge to inform and support a model that is trained on the sparse, high-fidelity confirmatory data. To tackle the noisy nature of the lower-fidelity measurements, we wish to develop models that can learn structure-activity relationships beyond simple molecular similarity and that can, ideally, learn smooth latent spaces structured according the measured activity.
Our approach is based on graph neural networks (GNNs), a class of algorithms that has been successfully applied for many molecular problems over the last few years. However, transfer learning is known to be difficult with GNNs, and for this reason it is also relatively understudied. To combat this, we leveraged a graph learning innovation that we recently introduced, in the form of adaptive readouts. The technique involves replacing a crucial part of GNNs, the readout function responsible for aggregating atom-level representations into a single molecular representation or embedding. Typically, GNN readouts are simple functions such as sum, mean, or maximum, while here we use an attention-based neural network for this purpose.
 presented schematically in a typical drug discovery scenario with a large and low-fidelity (LF) high-throughput screening dataset, and a sparse and high-fidelity (HF) confirmatory screening dataset")
In our work, we combine a variational graph autoencoder with adaptive readouts and show that the approach enables transfer learning on molecular data originating from drug discovery and quantum mechanics. Crucially, we show standard GNNs are not able to perform transfer learning for the challenging tasks we proposed. Our most striking result is that transfer learning as presented here can improve the predictive performance of confirmatory-level models by up to 8 times while using an order of magnitude less data. In our comprehensive evaluation, we tackled transductive and inductive settings, a multitude of protein targets and quantum properties, 3D atomic coordinate-aware networks like SchNet, and cases where more than 2 fidelities (i.e. low and high) are present in the data. Overall, we believe that transfer learning is the natural step forward in deep learning for molecular discovery. We envision several extensions, such as multi-task learning or generative capabilities, that can benefit from multi-fidelity data and can lead to an exciting future for drug discovery.
I am currently pursuing a PhD degree in Computer Science, with a focus on Machine Learning techniques for biology and chemistry. More generally, I am interested in applying Computer Science techniques in fields relevant to human biology and health, as well as rapidly developing areas like biotechnology, synthetic biology, and alternative computing paradigms.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in