Alzheimer’s disease modelling with human neurons reveals how amyloid cannot exert its toxicity without tau
Published in Microbiology, Neuroscience, and Cell & Molecular Biology
Explore the Research
Tau depletion in human neurons mitigates Aβ-driven toxicity - Molecular Psychiatry
Molecular Psychiatry - Tau depletion in human neurons mitigates Aβ-driven toxicity
The current understanding
Both Abeta and tau (referring to microtubule-associated protein tau) depositions in the brain are key pathologies that define Alzheimer’s disease (AD), with the former typically manifested prior to the latter. Multiple tau-deficient animal models have demonstrated that many aspects of Abeta-driven toxicity are dependent on the expression of tau, but how these pathological proteins interact in a human model remains unexplored. This is especially important when fundamental biological differences exist between mouse and human with regards to Alzheimer’s pathologies and tau expression patterns.
Multi-pronged strategies used to interrogate the tau-depleted cells
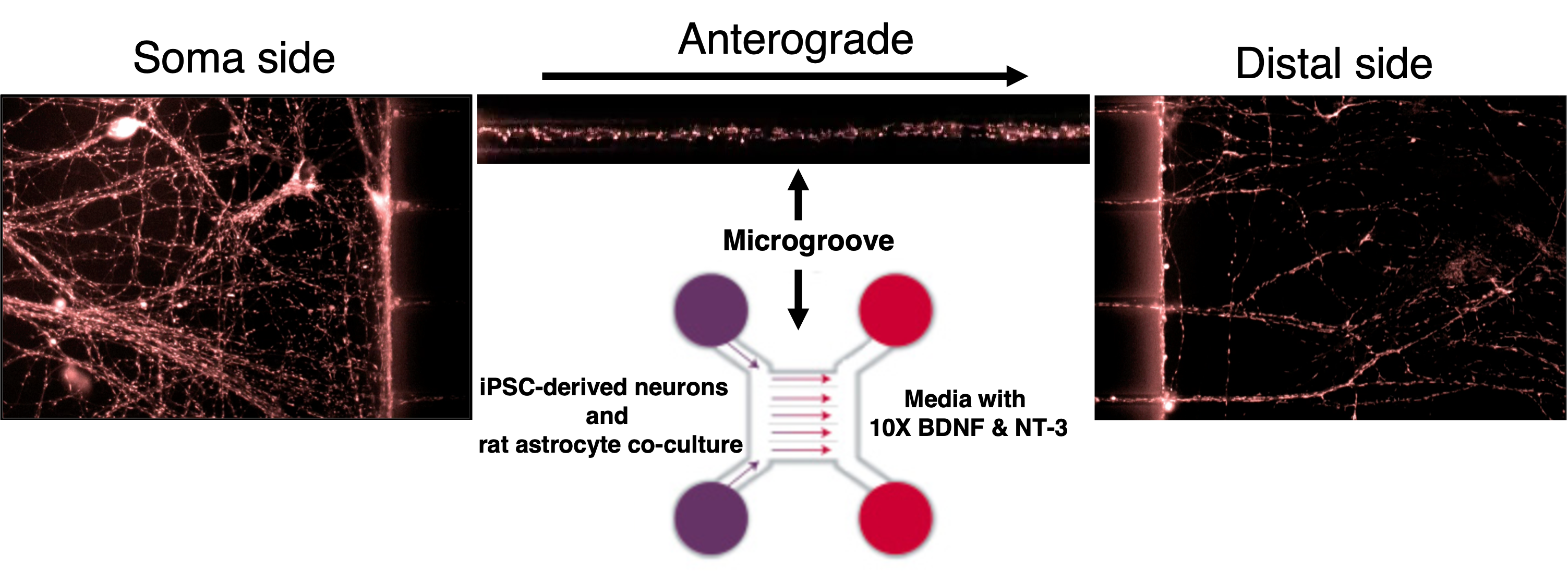
Working together with collaborating lab groups in Oxford, we used a CRISPR-Cas9 system to disrupt tau expression against the MAPT gene in two different loci and stem cell lines derived from two individuals to generate two isogenic panels for downstream experiments. The iPSCs were comprehensively validated by multiple assessments including DNA, RNA, and protein analyses (Figure 1). These edited iPSC lines were then turned into cortical neurons over 2-3 months before they displayed consistent neuronal activity levels. Subsequently, we treated the neurons with Abeta derived from different sources – synthetic and AD brain tissue-derived, before they were subject to a range of assays measuring from more sensitive parameters like neuronal activity to more drastic changes like cell death.
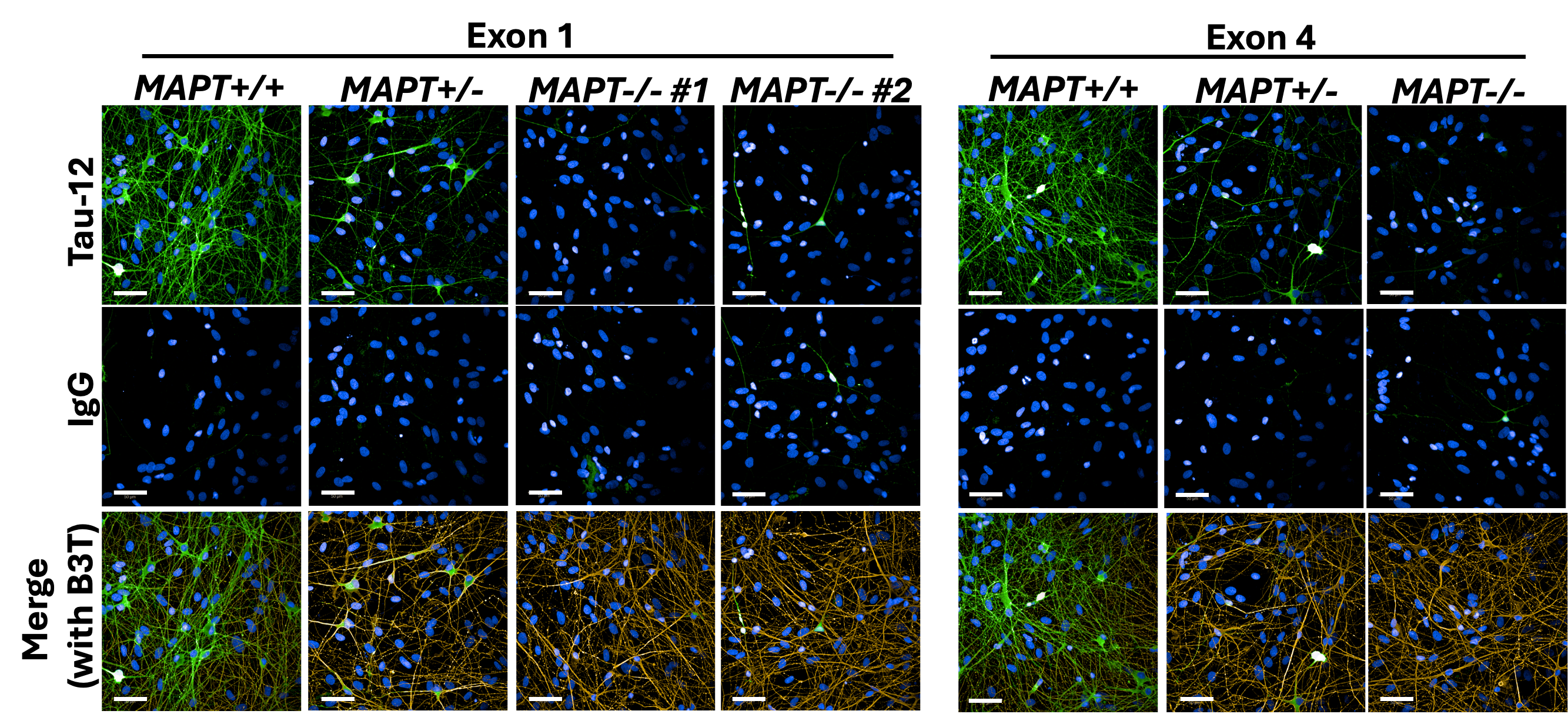
Figure 1: Immunocytochemistry demonstrating tau depletion in edited iPSC-derived cortical neurons (MAPT-/-) as compared to tau-expressing neurons (MAPT+/+) in two isogenic panels. Scale bar = 50 μm.
What the data showed us
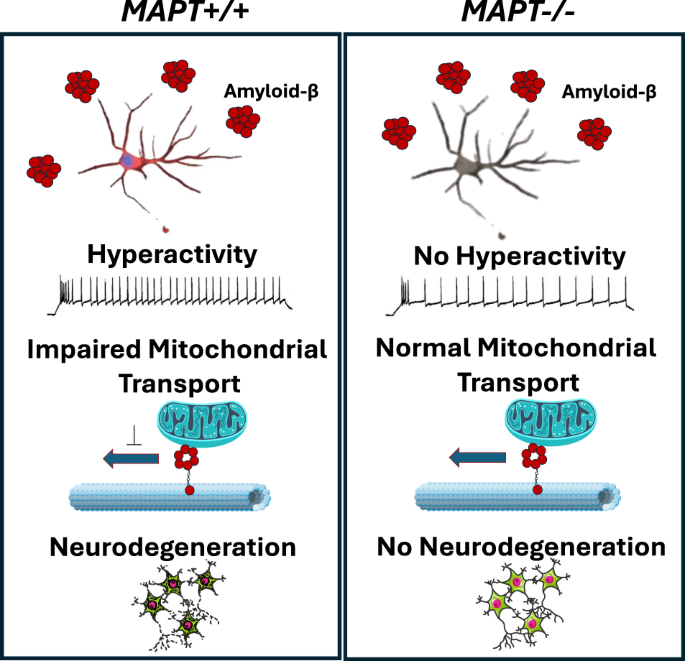
Without any Abeta insults, tau depletion caused significant reductions in neuronal activities and impairments of neurite outgrowth. Synaptic density, mitochondrial membrane potential and mitochondrial transport along axons remained unaffected by tau depletion in cortical neurons. After treating cortical neurons with Abeta, they exhibited hyperactivity, mitochondrial transport impairment and cell death which were all mitigated in tau-depleted neurons indicating that tau mediates Abeta-driven toxicity in these cellular phenotypes (Figure 2). Importantly, Abeta-driven cell death was also mitigated in heterozygous tau-depleted neurons (i.e. MAPT+/-; only one of the two alleles were disrupted by CRISPR) suggesting that partial reduction in tau expression levels is sufficient for protection.
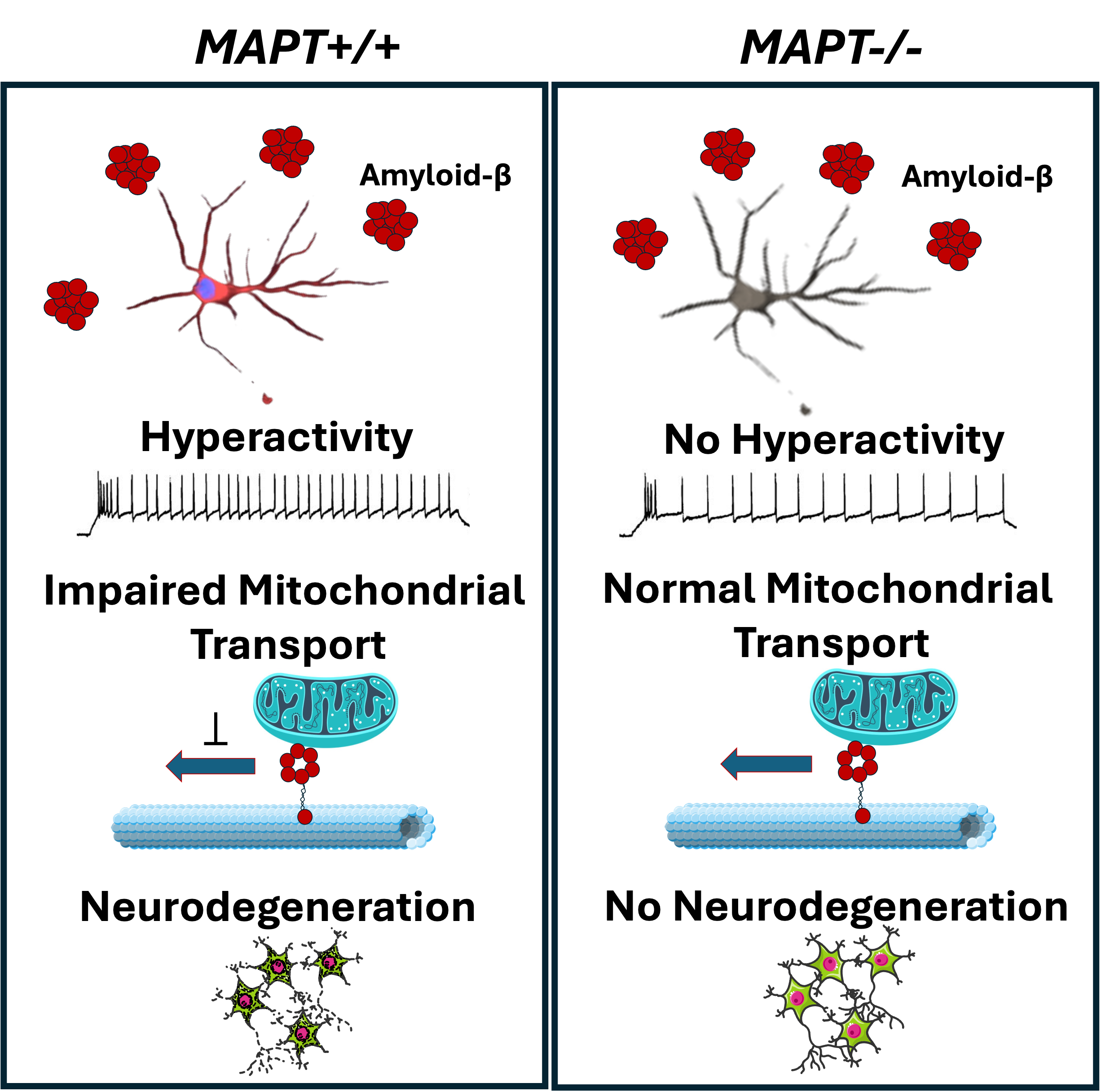
Figure 2: Graphical summary of Abeta-driven toxicity in tau-expressing and its mitigation in tau-depleted cortical neurons.
Why is it important?
Our study provided a valuable human model of tau depletion that is scalable (i.e. iPSCs proliferate and self-renew) and adaptable to other cell types (i.e. iPSCs can be differentiated into almost any cell types of interest in addition to cortical neurons shown in this study) whereas the data demonstrated that tau-dependency of Abeta-driven toxicity observed in animal models can be replicated in human neurons. This study is also highly relevant for multiple ongoing clinical trials employing tau lowering strategies – our data indicate that partial tau reductions strike a balance between mitigating Abeta-driven toxicity and causing cellular dysfunctions.
n.b. This study took place during my DPhil study at the University of Oxford, partly supported by the National Science Scholarship from the Agency for Science, Technology and Research (A*STAR) Singapore.
Follow the Topic
-
Molecular Psychiatry
This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in