Atomic High-Spin Cobalt(II) Center for Highly Selective Electrochemical CO Reduction to CH3OH
Published in Chemistry

Electrochemical CO2 reduction reaction (CO2RR), as a promising strategy for CO2 mitigation and transformation, has been extensively studied over the past few decades 1. Liquid products, especially CH3OH, possess significant advantages because of their high energy density and ease of storage 2. However, few catalysts are capable to convert CO2 to products other than CO or HCOOH with high activity and selectivity. Electrochemical CO reduction reaction (CORR) emerges as an encouraging approach to produce other products 3. Till now, developing efficient electrocatalysts for highly active and selective CORR remains challenging yet urgent.
Single atom catalysts (SACs) could act as a promising platform to selectively catalyze CORR due to the absence of consecutive active sites 4. However, few SACs have been reported to reduce CO to generate liquid products because SACs usually show weak adsorption towards *CO, which is detrimental to the subsequent hydrogenation reaction. Moreover, the real structure of experimentally synthesized SACs is rather complex that contains a large variety of single-atom centers with different coordination environments, which greatly complicates the understanding of the structure-performance relationship. On the other hand, molecular catalysts with precisely defined coordination structures offer a model system to probe the reaction mechanism and reveal the catalyst structure-performance relationship. Cobalt phthalocyanine (CoPc) molecular catalyst showed the capability to electrochemically reducing CO to CH3OH, however the current density and the CH3OH selectivity are still unsatisfactory. The spatial structure of binuclear CoPc is very different from that of mononuclear CoPc, which is anticipated to result in distinct crystal field and thus different catalytic performance, making binuclear CoPc an interesting candidate to be investigated in CORR.
In this work, Prof. Bin Liu’s group at City University of Hong Kong and Prof. Zhai’s group at Wuhan University proposes two model catalysts were constructed by anchoring cobalt phthalocyanine and binuclear cobalt phthalocyanine (M-CoPc and B-CoPc) on nitrogen doped carbon support. It is found that the B-CoPc can be transferred from low-spin state (LS, 1/2) to high-spin state (HS, 3/2) after thermal treatment, and the HS B-CoPc is able to catalyze CORR to methanol much more effectively than LS M-CoPc and B-CoPc. The CH3OH partial current density reaches about 35 mA/cm2 at -0.8 V (vs. RHE) with a methanol Faradaic efficiency (FE) of 50%. Operando attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) measurements and density functional theory (DFT) calculations disclose that the high spin Co2+ in HS B-CoPc can enable electron accumulation on *CO that will greatly weaken the C-O bonding, promoting the hydrogenation of CORR intermediates (*CO/*CHxO), which lead to boosted CORR activity and selectivity.
A series of characterization, including comparison of XRD, XPS, in-situ Raman, XAFS and HAADF-STEM. These results the structure of CoPc did not change significantly before and after the thermal treatment, and the first shell Co-N distance of B-CoPc-400 is slightly longer than that of B-CoPc-RT, which may be caused by molecular distortion induced by heat treatment.
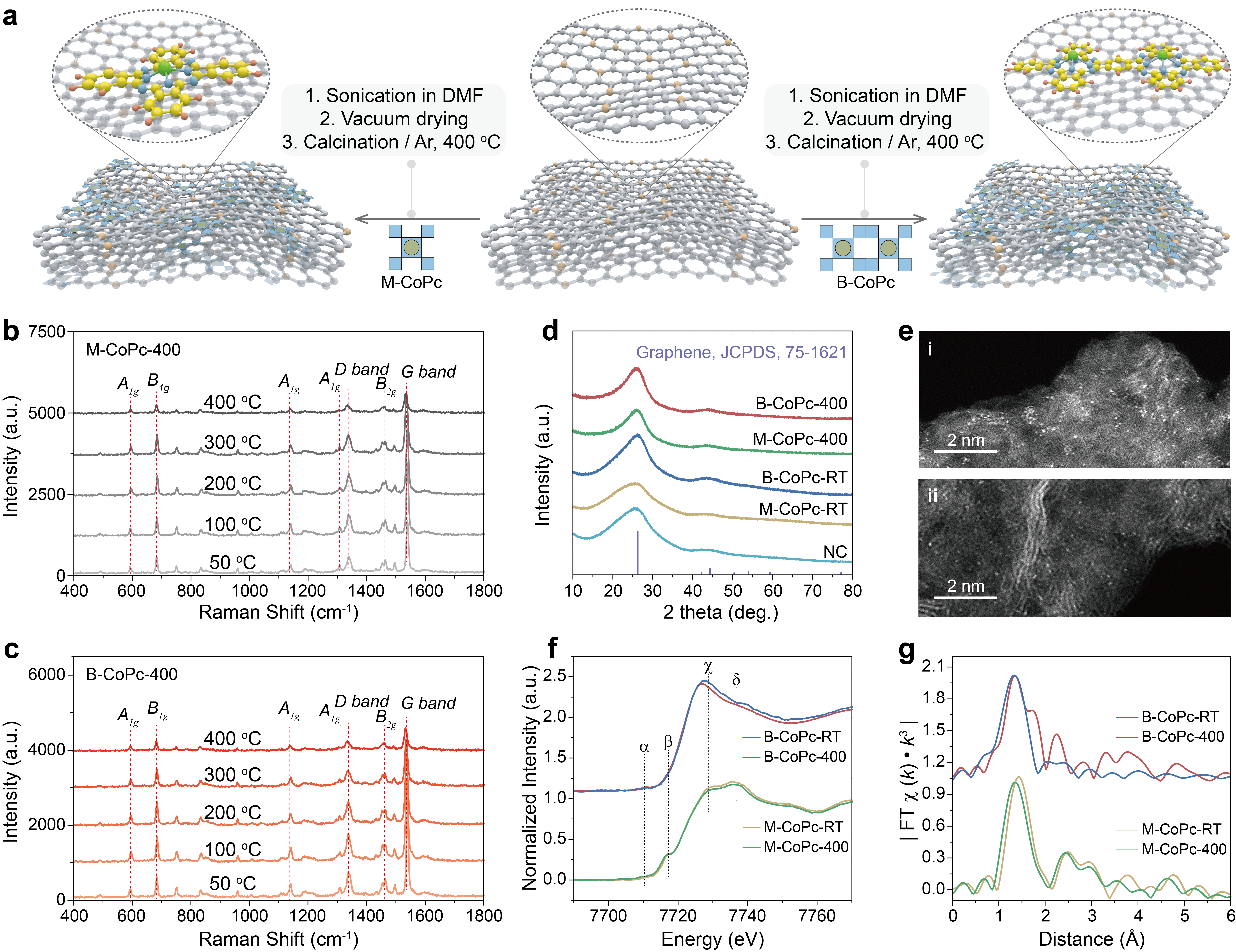 Figure 1 Structural characterization. (a) Schematic of the synthesis process for M-CoPc-RT/400 and B-CoPc-RT/400. In-situ Raman spectra of (b) M-CoPc-RT and (c) B-CoPc-RT recorded during the thermal treatment process. (d) X-ray diffraction patterns. (e) The high magnification HAADF-STEM image of (i) M-CoPc-400 and (ii) B-CoPc-400 (scale bar, 2 nm). (f) Co K-edge XANES spectra before and after annealing. (g) The corresponding Fourier transformation (FT)-EXAFS spectra.
Figure 1 Structural characterization. (a) Schematic of the synthesis process for M-CoPc-RT/400 and B-CoPc-RT/400. In-situ Raman spectra of (b) M-CoPc-RT and (c) B-CoPc-RT recorded during the thermal treatment process. (d) X-ray diffraction patterns. (e) The high magnification HAADF-STEM image of (i) M-CoPc-400 and (ii) B-CoPc-400 (scale bar, 2 nm). (f) Co K-edge XANES spectra before and after annealing. (g) The corresponding Fourier transformation (FT)-EXAFS spectra.
The two thermally treated catalysts display much higher current densities than the untreated ones. Additionally, the linear sweep voltammetry (LSV) curves (Figure 2b) further show that the B-CoPc-400 exhibits a much better CORR performance than M-CoPc-400. Figure 2c compares the potential-dependent product distribution of CORR over M-CoPc-400 and B-CoPc-400. The main product on M-CoPc-400 is H2, with a Faradaic efficiency (FE) over 80% in the potential range from -0.5 V to -1.0 V (vs. RHE). The maximum FE for CH3OH over M-CoPc-400 is ~19 % at -0.7 V (vs. RHE), matching well with the early work. Moreover, the B-CoPc-400 could produce CH3OH with a FE of 53% at -0.7 V (vs. RHE) with negligible current loss for 10 h, corroborating its excellent catalytic durability (Figure 2d).
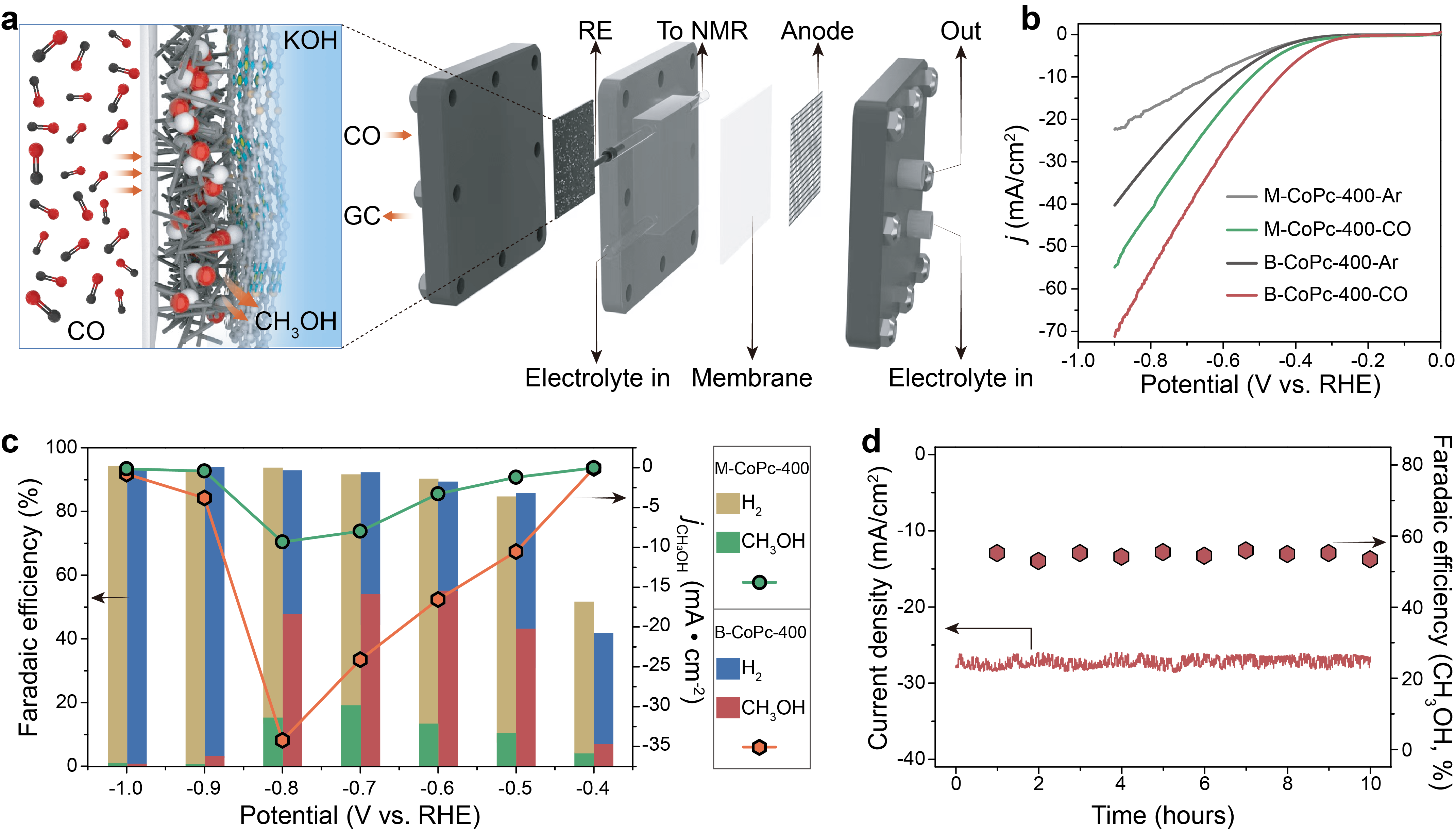 Figure 2 Catalytic performance. (a) Schematic diagram showing the MEA for CORR. (b) LSV curves acquired in Ar and CO-saturated 0.5 M KOH solution on a carbon paper at a scan rate of 5 mV/s. (c) Potential-dependent product selectivity for CORR catalyzed by M-CoPc-400 and B-CoPc-400. (d) Stability of M-CoPc-400 recorded at -0.7 V (vs. RHE). The measurement was performed at the condition of 1 atm CO and room temperature in CO-saturated 0.5 M KOH.
Figure 2 Catalytic performance. (a) Schematic diagram showing the MEA for CORR. (b) LSV curves acquired in Ar and CO-saturated 0.5 M KOH solution on a carbon paper at a scan rate of 5 mV/s. (c) Potential-dependent product selectivity for CORR catalyzed by M-CoPc-400 and B-CoPc-400. (d) Stability of M-CoPc-400 recorded at -0.7 V (vs. RHE). The measurement was performed at the condition of 1 atm CO and room temperature in CO-saturated 0.5 M KOH.
To understand the different CORR catalytic activity as well as the CORR mechanism over M-CoPc-400 and B-CoPc-400, operando ATR-SEIRAS was conducted to probe the reaction intermediates. To further examine the relation between kinetics of CORR and proton-feeding, the kinetic isotope effect (KIE) of H/D was measured using H2O and D2O as the proton sources, which was performed at -0.6 V (vs. RHE). (Figure 3) From the operando ATR-SEIRAS and KIE results, it can be deduced that the CORR over M-CoPc-400 and B-CoPc-400 was rate-limited by the *CO/*CHxO hydrogenation and methanol desorption step, respectively.
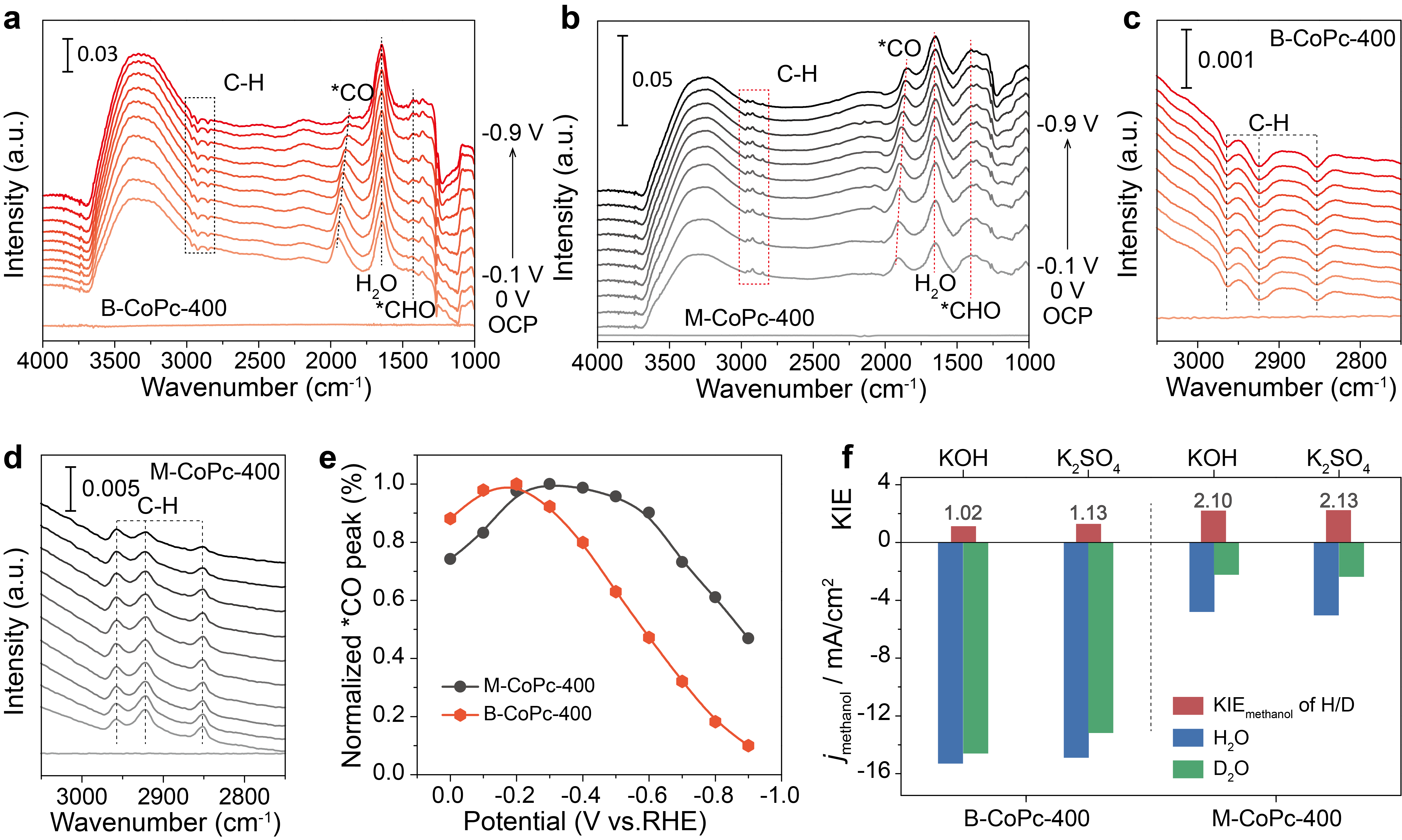 Figure 3 Operando ATR-SEIRAS and KIE measurements. Operando ATR-SEIRAS spectra of CO reduction over (a, c) B-CoPc-400 and (b, d) M-CoPc-400 in CO-saturated 0.5 M KOH. The spectra were collected at constant potentials with 0.1 V interval in the cathodic direction from OCP to -0.9 V (vs. RHE). (e) Potential dependent CO stretching peak intensity. (f) KIE of H/D in CORR to CH3OH at -0.6 V (vs. RHE) over B-CoPc-400 and M-CoPc-400.
Figure 3 Operando ATR-SEIRAS and KIE measurements. Operando ATR-SEIRAS spectra of CO reduction over (a, c) B-CoPc-400 and (b, d) M-CoPc-400 in CO-saturated 0.5 M KOH. The spectra were collected at constant potentials with 0.1 V interval in the cathodic direction from OCP to -0.9 V (vs. RHE). (e) Potential dependent CO stretching peak intensity. (f) KIE of H/D in CORR to CH3OH at -0.6 V (vs. RHE) over B-CoPc-400 and M-CoPc-400.
To dig out the origin of fast rate of hydrogenation over B-CoPc-400 in CORR to methanol, Co L2,3-edge XAS and EPR measurements were conducted to differentiate the electronic structure between M-CoPc-400 and B-CoPc-400. These results suggest that the 400 °C treatment of B-CoPc-RT changes its spin structure from low spin to high spin.
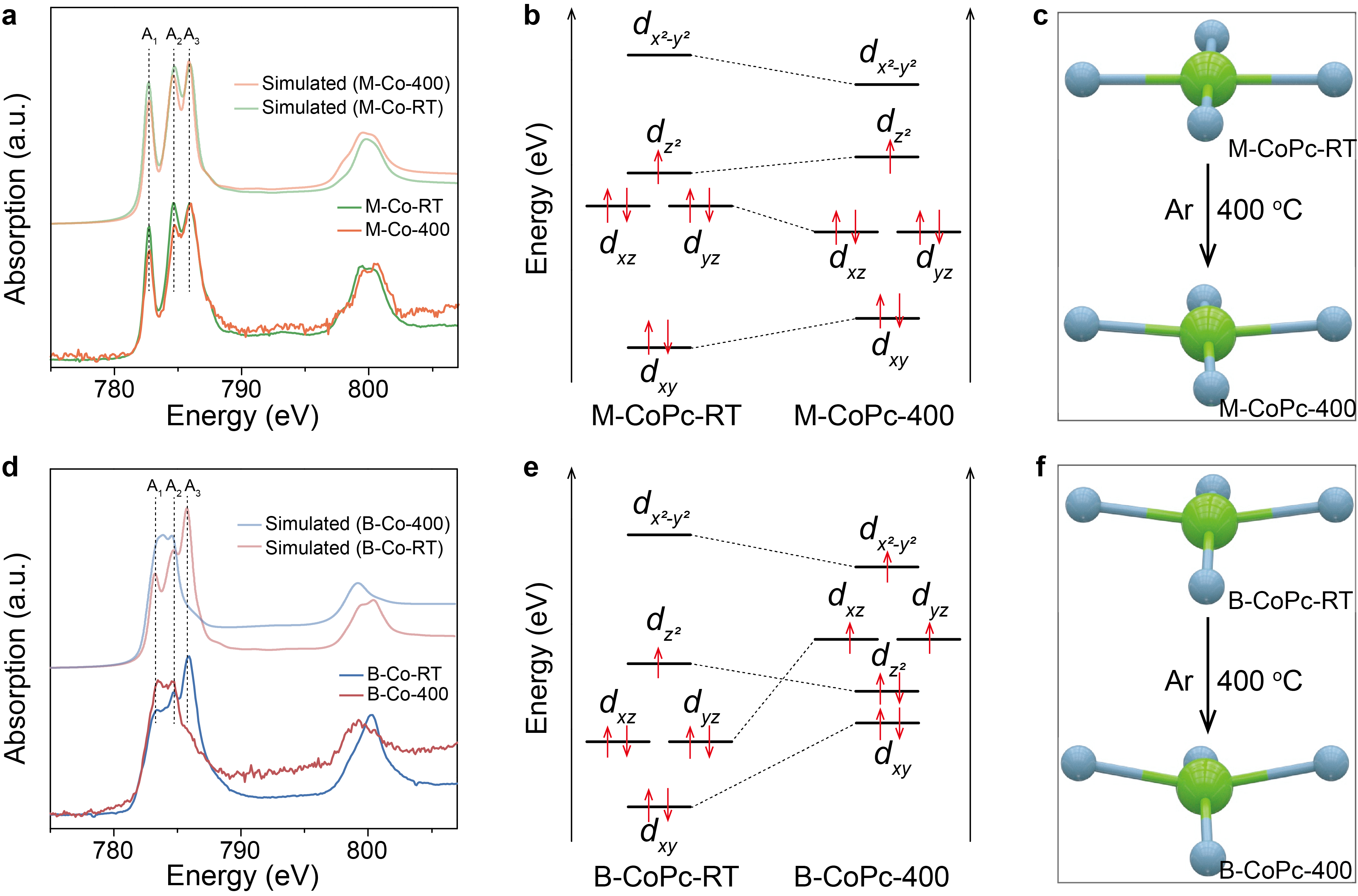 Figure 4 Analysis of electronic structure. (a, d) The experimental and simulated cobalt L2,3-edge XAS spectra of M-CoPc-RT/400 and B-CoPc-RT/400, respectively. (b, e) 3d orbital diagrams of M-CoPc-RT/400 and B-CoPc-RT/400, respectively. (c, f) Schematic showing the effect of thermal treatment on the coordination environment of M-CoPc-RT/400 and B-CoPc-RT/400, respectively.
Figure 4 Analysis of electronic structure. (a, d) The experimental and simulated cobalt L2,3-edge XAS spectra of M-CoPc-RT/400 and B-CoPc-RT/400, respectively. (b, e) 3d orbital diagrams of M-CoPc-RT/400 and B-CoPc-RT/400, respectively. (c, f) Schematic showing the effect of thermal treatment on the coordination environment of M-CoPc-RT/400 and B-CoPc-RT/400, respectively.
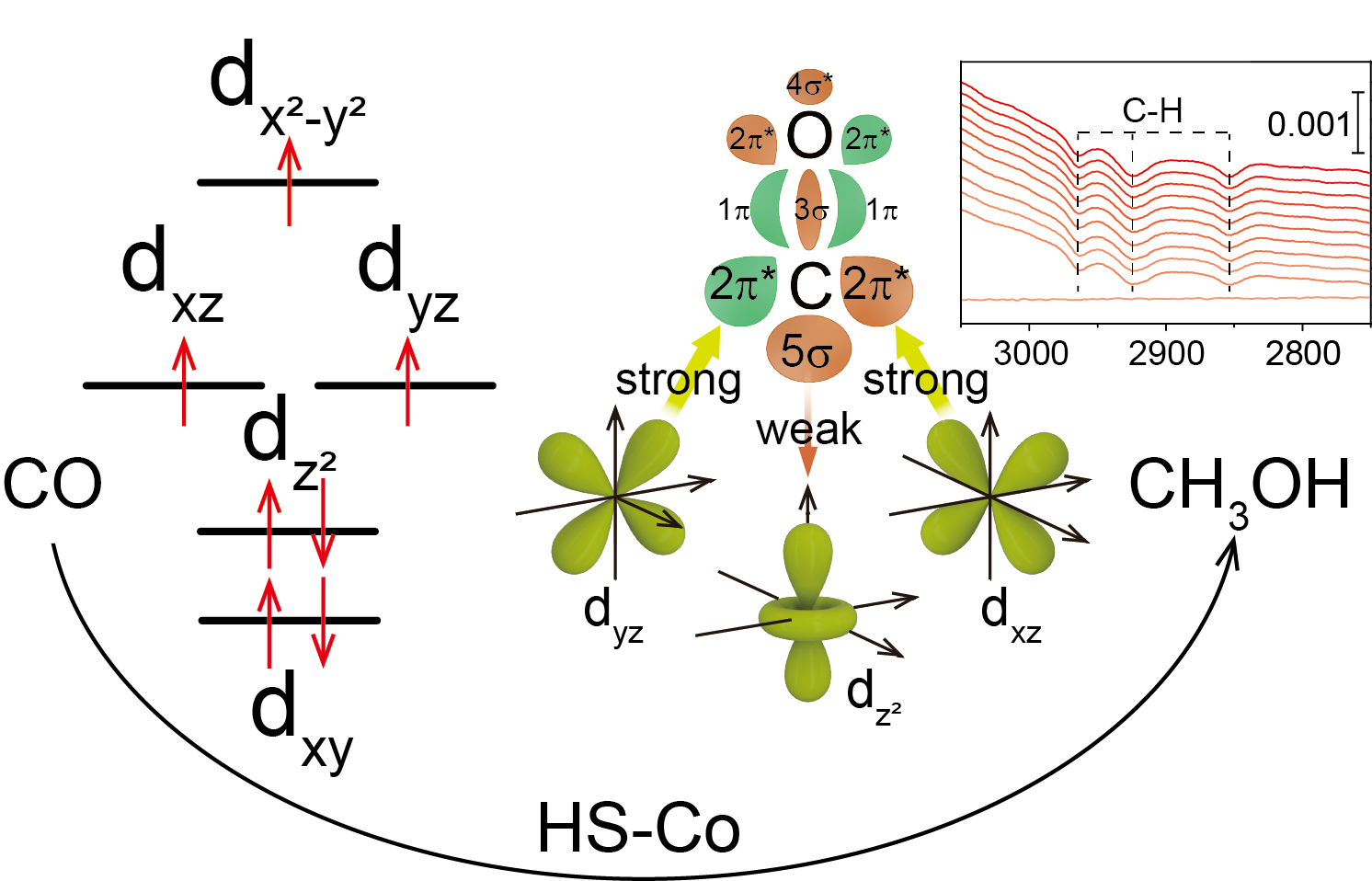
To further clarify the effect of Co spin state on CORR performance, DFT calculation was performed to compare the reaction free energy of CORR on CoPc and distorted CoPc. The results show the schematic of σ and π-donation bonds between CO and 3d orbitals of LS-Co2+-0° and HS-Co2+-15°; the weaker 3dz2-5σ and more electron transfer from Co to *CO via π back-donation for HS-Co2+-15° lead to smaller nCT from CO to Co site, enabling more electron accumulation on the 2π* orbital of *CO, which will effectively promote the hydrogenation of CO reduction intermediates ca. *CO (similar to *OCH2) in CORR. Once *CO formation on Co sites occurs, under cathodic bias, the electrons from the electrode will fill into the highest unpaired orbitals of Co sites, 3dz2 and 3dxz/dyz for LS-Co2+-0° and HS-Co2+-15°, respectively. In the case of LS-Co2+-0°-CO, the electrons from the electrode will fill in the anti-bonding orbital of 3dz2-5σ bond, resulting in weakened *CO adsorption strength, unfavorable for *CO reduction. While in the case of HS-Co2+-15°-CO, the electrons from the electrode will fill in the bonding orbital of 3dxz/dyz-2π* bond, weakening the C-O bond in *CO, which can effectively promote *CO hydrogenation. (Figure 5)
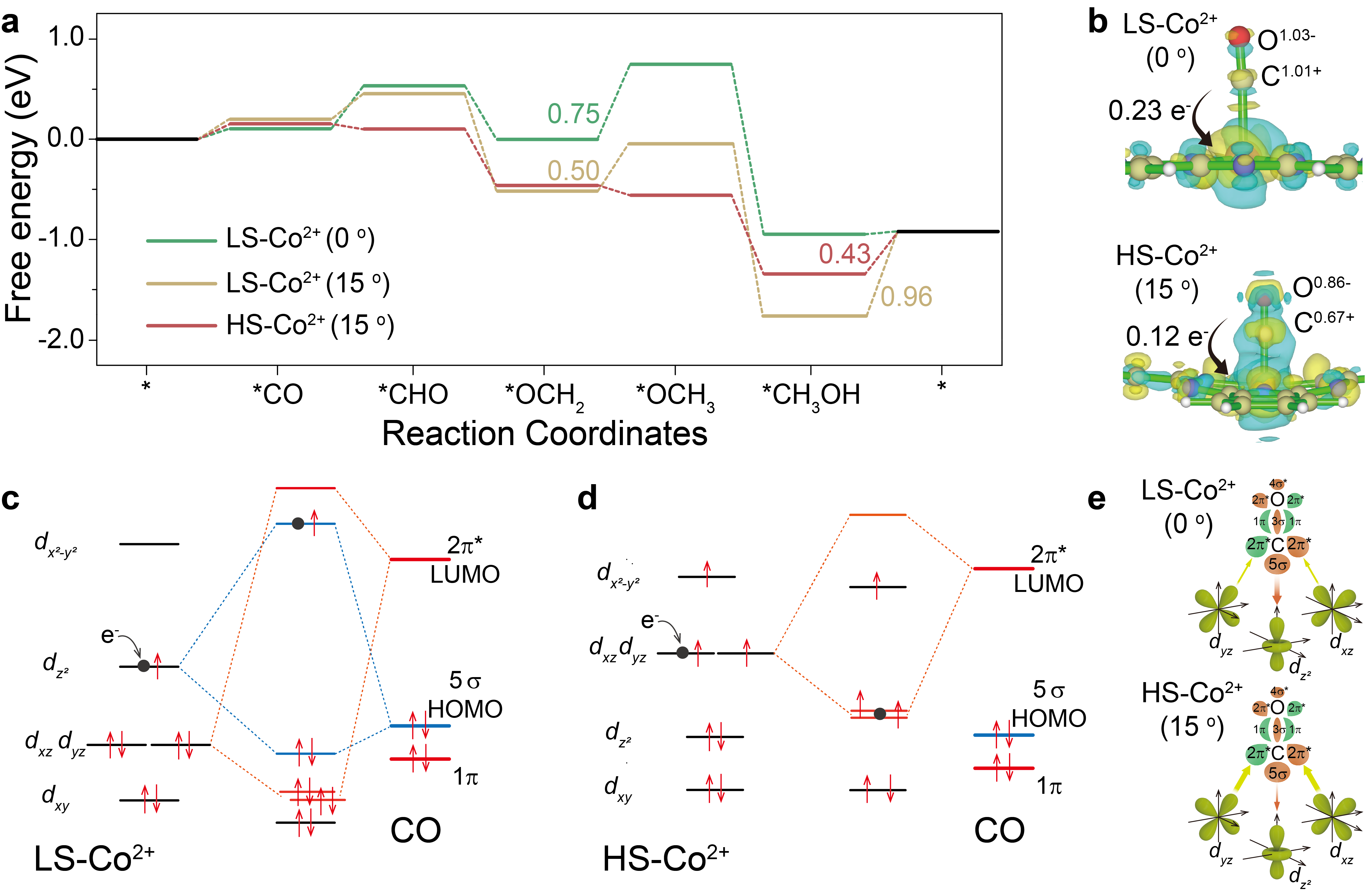 Figure 5 CORR mechanism. (a) Free energy diagram of CORR over LS-Co2+-(0 and 15°) and HS-Co2+-(15°). (b) The charge density distributions of LS-Co2+-CO and HS-Co2+-CO. Blue, yellow, pink and gray balls represent N, C, Co and O atoms, respectively, and the faint yellow and cyan regions refer to the increased and decreased charge density. Interactions between CO molecular frontier orbitals (5σ and 2π*) and the 3d orbital of (c) LS-Co2+ and (d) HS-Co2+ site. Dot in black of (c) and (d) is the electron from electrode under cathodic bias. (e) Schematic of σ and π-donation bonds between CO and 3d orbital of LS-Co2+ and HS-Co2+. The size of the arrow indicates the binding strength of 3dz2-5σ (in red color) and 3dxz/dyz-2π* (in green color), which also indicates the magnitude of the electron migration.
Figure 5 CORR mechanism. (a) Free energy diagram of CORR over LS-Co2+-(0 and 15°) and HS-Co2+-(15°). (b) The charge density distributions of LS-Co2+-CO and HS-Co2+-CO. Blue, yellow, pink and gray balls represent N, C, Co and O atoms, respectively, and the faint yellow and cyan regions refer to the increased and decreased charge density. Interactions between CO molecular frontier orbitals (5σ and 2π*) and the 3d orbital of (c) LS-Co2+ and (d) HS-Co2+ site. Dot in black of (c) and (d) is the electron from electrode under cathodic bias. (e) Schematic of σ and π-donation bonds between CO and 3d orbital of LS-Co2+ and HS-Co2+. The size of the arrow indicates the binding strength of 3dz2-5σ (in red color) and 3dxz/dyz-2π* (in green color), which also indicates the magnitude of the electron migration.
In this work, Dr. Jie Ding, Prof. Yueming Zhai, Prof. Bin Liu and co-authors found that L-edge XAS analysis on a model single-Co-atom catalyst verified the transformation of single atomic cobalt center from LS (S=1/2) to HS (S=3/2) after thermal treatment, which greatly promoted CORR performance. Combining operando ATR-SEIRAS, KIE experiments and DFT calculations, it showed that the change of spin state of single atomic cobalt center (II) from LS (S=1/2) to HS (S=3/2) resulted in a change of the CORR RDS from *CO/*CHxO hydrogenation to methanol desorption, corresponding to a decrease of the RDS energy barrier by 0.32 eV from 0.75 to 0.43 eV. This work indicates that the chemical bonding of CO molecule on single atom catalyst determined by the specific orbital characters of d-bands is critical for the hydrogenation process in CORR, which provides valuable information for understanding the electrochemical catalytic reaction at the molecular level.
References
1. Ding, J. et al. Circumventing CO2 reduction scaling relations over the heteronuclear diatomic catalytic pair. J. Am. Chem. Soc. 145, 11829-11836. (2023).
2. Navarro-Jaén, S. et al. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 5, 564-579 (2021).
3. Ren, X. et al. In-situ spectroscopic probe of the intrinsic structure feature of single-atom center in electrochemical CO/CO2 reduction to methanol. Nat. Commun. 14, 3401 (2023).
4. O’Mara, P. B. et al. Cascade reactions in nanozymes: Spatially separated active sites inside Ag-Core–Porous-Cu-shell nanoparticles for multistep carbon dioxide reduction to higher organic molecules. J. Am. Chem. Soc. 141, 14093-14097 (2019).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in