Automatically Determining Catalytic Reaction Mechanisms Using High-Throughput Deep Reinforcement Learning with First Principles
Published in Chemistry and Computational Sciences
Understanding catalytic reactions is essential for improving chemical processes, refining reaction conditions, and developing effective catalysts. Knowing the reaction mechanisms helps create more selective and efficient catalysts, optimizing their design, reducing side reactions, and enhancing selectivity by controlling activation energy. Additionally, understanding optimal reaction conditions like temperature, pressure, and reactant concentration can boost yield and efficiency. However, exploring reaction pathways in catalytic reactions is challenging due to the complexity of multi-step reactions, short-lived intermediates, and the dynamic nature of many catalytic reactions.
Recently, Artificial Intelligence (AI) has become a vital tool in various research fields, including chemistry. Although machine learning algorithms have been applied to chemical reactivity, most are mechanism-agnostic and require human interpretation. Reinforcement Learning (RL) offers substantial potential for exploring reaction networks and mechanisms by allowing an agent to identify plausible pathways through interactions with a defined environment. This approach can automate the exploration of reaction networks.
 framework for studying hydrogenation reactions in ammonia synthesis on the Fe(111) surface.")
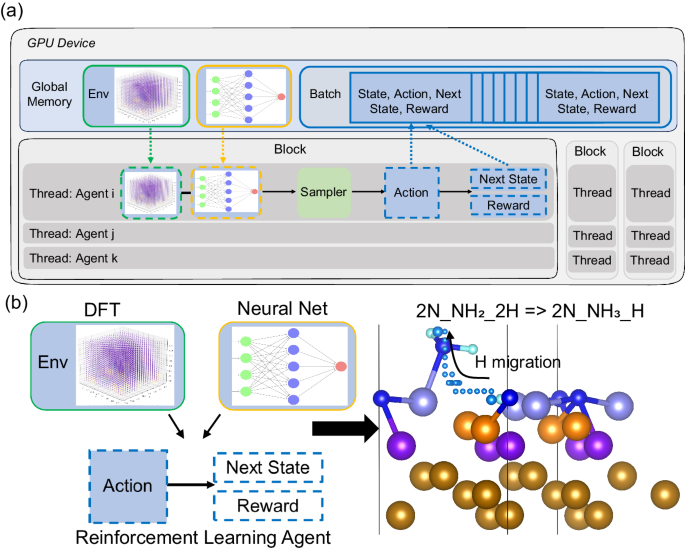
Figure 1. High-Throughput deep reinforcement learning (RL) framework for studying hydrogenation reactions in ammonia synthesis on the Fe(111) surface.
In this work, the authors, Dr. Tian Lan at Salesforce A.I. Research and Prof. Qi An at Iowa State University, developed a new framework called high-throughput deep reinforcement learning (HDRL-FP). HDRL-FP overcomes the limitations of previous methods by being reaction-agnostic and high-throughput. It operates on atomic positions mapped to the potential energy landscape from the first principles. This allows for thousands of concurrent RL simulations on a single GPU, improving training stability and reducing runtime. HDRL-FP’s effectiveness was demonstrated by predicting reaction paths in the Haber-Bosch process, showing its potential to optimize chemical reactions and reduce costs.
The HDRL-FP framework, displayed in Figure 1, revolutionizes the RL workflow by executing it end-to-end on a single GPU. This approach leverages the parallelization capabilities of GPUs, enabling cost-efficient and high-throughput computation. HDRL-FP runs thousands of concurrent RL simulations in parallel, training on large batches of experience efficiently. Within the GPU, numerous replicas of environment instances operate independently, each maintaining its own reference of the potential energy landscape (PEL) computed by density functional theory (DFT) and shared deep policy models. This setup allows HDRL-FP to handle complex chemical reactions. Each environment instance supports multiple concurrent threads, with atom actors sampling actions, transitioning to new states, and collecting rewards. Experience steps are stored in the global GPU memory, ensuring seamless data handling. HDRL-FP automatically resets completed environments without interrupting ongoing ones. Once experience data is gathered, training on large batches synchronizes atom actors for cohesive and faster convergence.
 on Fe(111) surface, differentiated by Langmuir-Hinshelwood (LH) and Eley-Rideal (ER) mechanisms.")
Figure 2. Hydrogen migration pathways and potential energy landscape (PEL) on Fe(111) surface, differentiated by Langmuir-Hinshelwood (LH) and Eley-Rideal (ER) mechanisms.
Next, HDRL-FP was applied to construct the reaction path for the crucial hydrogenation step in NH3 synthesis on the Fe(111) surface, transitioning from 2N_NH2_2H to 2N_NH3_H. This step is significant as it represents a major rate-determining process in the Haber-Bosch reaction network. HDRL-FP’s ability to handle thousands of concurrent RL simulations on a single GPU enabled efficient exploration of these mechanisms. The framework demonstrated robust convergence, especially with 100 and 500 environment replicas, highlighting the importance of massive parallelism in RL for exploring complex reaction mechanisms.
Figure 2 shows the results. Figure 2(a) displays the hydrogen migration paths for both the Langmuir-Hinshelwood (LH) and Eley-Rideal (ER) mechanisms as identified by HDRL-FP. Figures 2(b) and 2(c) provide detailed views of the atomic configurations for each mechanism. In the LH mechanism, the hydrogen atom moves laterally across the surface, navigating high-energy areas due to other atoms, before moving upwards towards NH2. In the ER mechanism, the hydrogen atom gradually descends to its final state. Figure 2(d) illustrates the energy paths, revealing an intermediate lower-energy state discovered by HDRL-FP and confirmed by further calculations, highlighting significant findings in hydrogen migration.
In summary, HDRL-FP is an AI framework for autonomously mapping complex catalytic reaction paths. This method avoids the need for empirical design of RL environments by relying on PEL derived from first principles and atomic positions. HDRL-FP’s high-throughput capability ensures rapid convergence and training stability across diverse catalytic reactions, reducing the need for hyperparameter fine-tuning. It adapts to various reaction complexities and works well in both non-relaxation and constrained relaxation environments. Future research will focus on combining HDRL-FP with constraint design to explore catalytic reaction mechanisms in more complex environments.
Starting from our 2021 JACS work (J. Am. Chem. Soc. 143, 16804–16812, 2021), we integrated all the code into the GPU using CUDA, boosting efficiency by 1000 times and significantly enhancing the convergence of reinforcement learning. This approach eliminated all approximations, transforming the search for chemical reaction pathways into a standardized reinforcement learning task of optimizing paths on a large, noisy 3D grid. For more details, please refer to the full research article at https://doi.org/10.1038/s41467-024-50531-6.
Dr. Qi An is an Associate Professor in the Department of Materials Science and Engineering at the Iowa State University. He received his MS and Ph.D. degrees in Materials Science from Caltech, and his BS degree from the University of Science and Technology of China. His research area is computational materials science.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in