Breaking linear scaling relationships in oxygen evolution via dynamic structural regulation of active sites
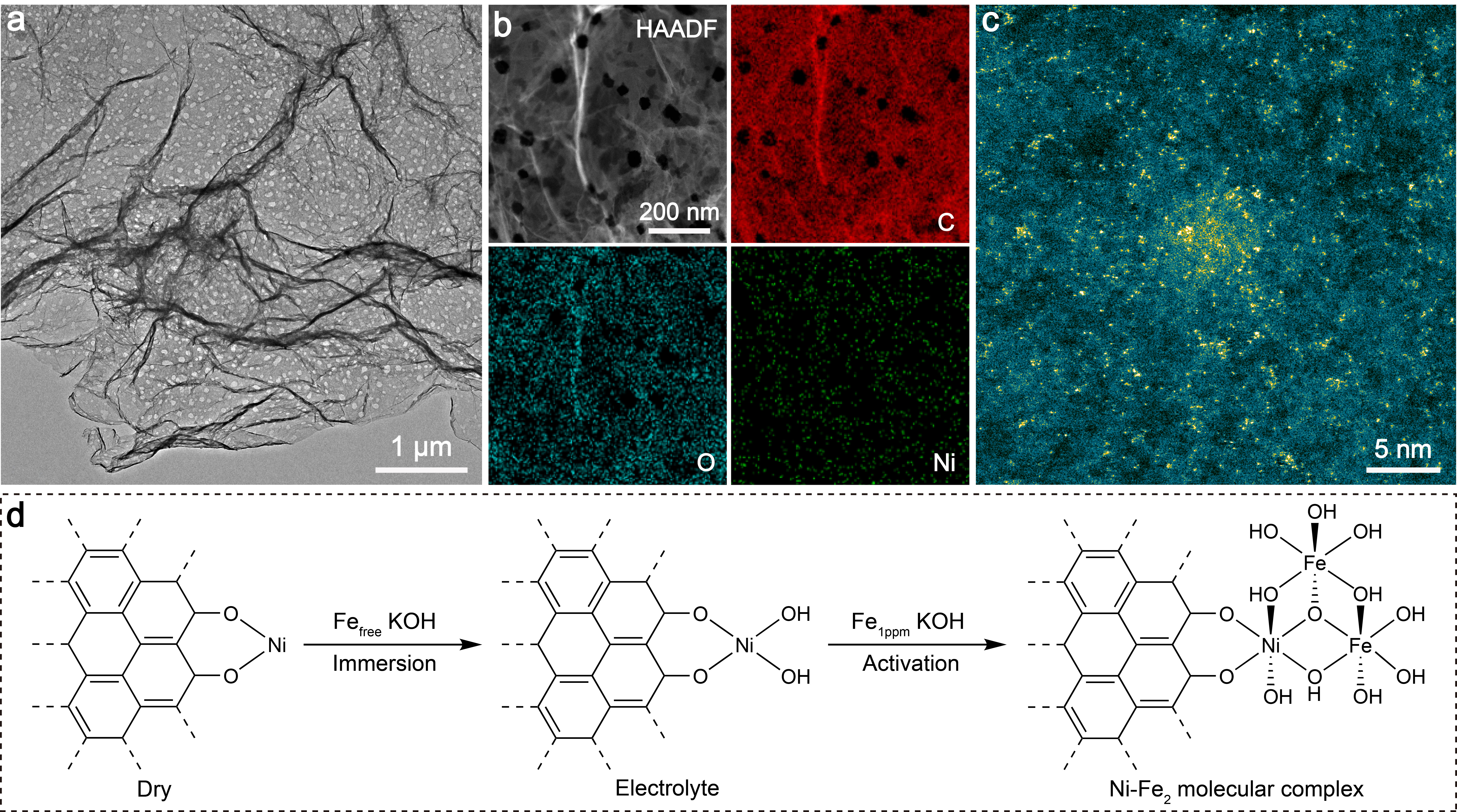
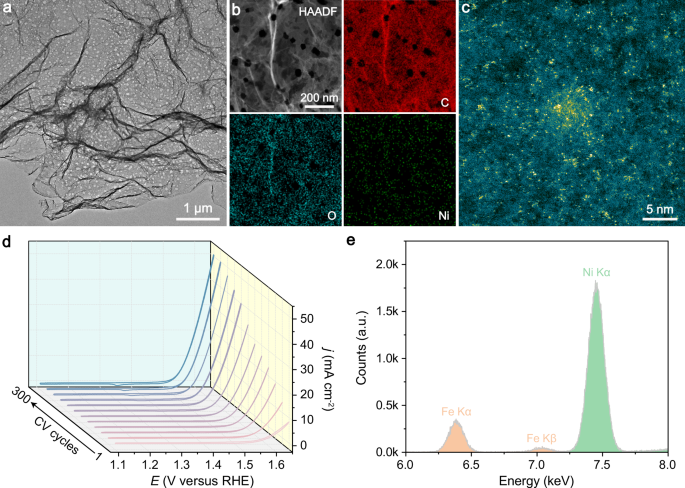
For multi-step catalytic reactions, it has been shown that the adsorption energies of different intermediates are correlated on conventional single-site catalysts by linear scaling relationships (LSRs). While LSRs simplify the prediction of catalyst performance and help elucidate catalytic activity trends, they inevitably place intrinsic limitations on optimally adjusting adsorption of every intermediate on the catalytic site simultaneously to achieve the maximum activity and/or selectivity. A case in point of this correlation-imposed reaction is the electrochemical oxygen evolution reaction (OER), which serves as an ideal anodic reaction by providing electrons and protons for hydrogen generation, CO2 reduction, etc. The adsorption energies of the chemically similar oxygenated intermediates over a single active site, such as *OH, *O, and *OOH involved in OER with the widely accepted adsorbate evolution mechanism (AEM), are linearly correlated and cannot be adjusted independently. Such correlation-imposed constraint makes it rather challenging for the maximal accomplishable catalytic performance.
On account of the ubiquitous adsorption-energy scaling relationship between *OOH and *OH on various OER electrocatalysts, one of the most effective strategies to break such LSR is to selectively stabilize *OOH over *OH by confining intermediates within nanoscopic channels, introducing proton acceptors, and/or creating multifunctional surfaces and interfacial sites . These strategies share a common idea of engineering heterogeneity of electrocatalysts. Nevertheless, atomistic insights into complex multiphase materials or interfaces during OER are very limited and remain elusive. Specifically, few experimental attempts have been made using heterostructured catalysts, but their heterogeneous nature poses great challenges in refining the structures of the most active surficial catalytic sites for use as the synthetic targets. Furthermore, static structures identified through ex situ characterization cannot accurately reflect the real active sites responsible for catalytic activity under reaction conditions. These limitations greatly impede the understanding of structure–performance relationships and the rational design of next generation high-performance OER electrocatalysts.
While structure changes in catalysts under electrochemical conditions have been extensively observed, the underlying coordination evolution of active sites within a catalytic cycle and the associated intrinsic electronic origin are often overlooked. Recent studies have shown that dynamic evolution of metal-support coordination during catalysis can effectively alter the local electronic structure of the active centre. This dynamic regulation of the active centre enables simultaneous facilitation of reactant activation and product desorption, thereby circumventing the scaling relations in heterogeneous catalysis. Therefore, exploring and comprehending the local coordination evolution of active sites at the atomic scale within the OER catalytic cycle could offer the opportunity to circumvent the existing scaling relationships. Given that bimetallic Ni-Fe composites have been identified as one of the most promising OER catalysts in alkaline conditions, they can serve as an ideal platform to investigate the above hypothesis.
In this work, we report the construction of a Ni-Fe2 molecular complex catalyst, which exhibits a notable OER performance. The catalyst preparation is based on in situ electrochemical activation of a low‐coordinate Ni single-atom pre-catalyst in purified KOH electrolyte with a deliberate addition of Fe ions at ppm level. Operando X-ray absorption fine structure (XAFS) measurements verify the structural transformation from Ni monomer to O-bridged Ni-Fe2 trimer during the activation process. Remarkably, by employing density functional theory (DFT) combined ab initio molecular dynamics (AIMD) simulations and electrokinetic characterization, we report an unconventional dynamic dual-site-cooperated OER mechanism, in which the Ni centre directly participates in the catalytic process to induce intramolecular proton transfer and trigger its coordination evolution. The existence of dynamic coordination between the Ni site and adsorbates (OH and H2O) plays a key role in modulating the electronic structure of the adjacent Fe active site during the OER cycle. Such dynamic regulation contributes to lowering the free energy required for the mutually competing steps of O–H bond cleavage and *OOH formation simultaneously, which effectively surmounts the LSRs in OER. Our insights into dynamic structural evolution of active site within the catalytic cycle enlighten a path towards the development of highly efficient OER catalysts beyond the limitations of LSRs.
For more details on the experiments and results, please refer to our paper published in Nature Communications: https://www.nature.com/articles/s41467-024-55150-9
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in