Cancer, Translation Stress and NPM1, a New Cancer Vulnerability
Published in Cancer and Cell & Molecular Biology
The variability in genetic mutations that drive tumours in different tissues is quite remarkable. Why mutations with clear oncogenic potential fail to cause cancer anywhere in the body is a real head-scratcher, and one we set out to explore. It has been noted that different tissues have distinct signalling requirements to permit tumour formation, but the extent of this fine-tuned balance is quite extraordinary! Even within the same signalling pathway, such as the WNT pathway that we study, different components can be differentially required for tumor development depending on the tissue. For instance, the WNT pathway component adenomatosis polyposis coli (APC) is mutated in ~80% of colorectal cancers (CRC), but rarely altered in hepatocellular carcinomas (HCC; ~1.2-5.5%). Conversely, mutations in the WNT effector β-catenin (CTNNB1) are found in ~20–40% of HCCs, but occur in only ~1.9% of CRCs, despite both types of mutations resulting in overactivation of the same pathway.
The question
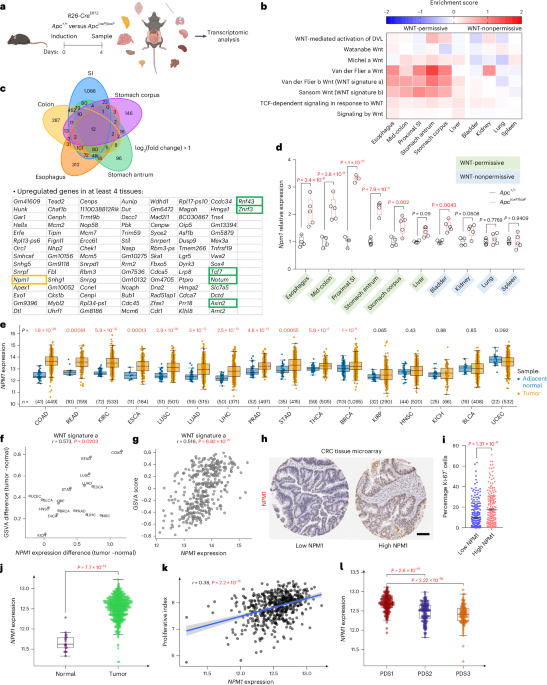
If we delete the tumour suppressor Apc, this potent WNT modulator mutated in the vast majority of CRCs, throughout the entire body, can we identify a set of genes that are consistently altered across most affected tissues? The goal: to uncover a shared, actionable target that could ultimately be translated into a therapeutic benefit for patients.
The target
The tissues most affected by this perturbation were those along the gastrointestinal tract. Surprisingly, despite the shared genetic alteration, only a small number of genes were consistently deregulated across all these tissues. Among them were well-known regulators of the WNT pathway, along with a few intriguing novel candidates. Given what we already know about the importance of MYC downstream of APC loss in driving WNT-mediated transformation, one particular gene caught our attention: nucleophosmin (NPM1).
The human relevance
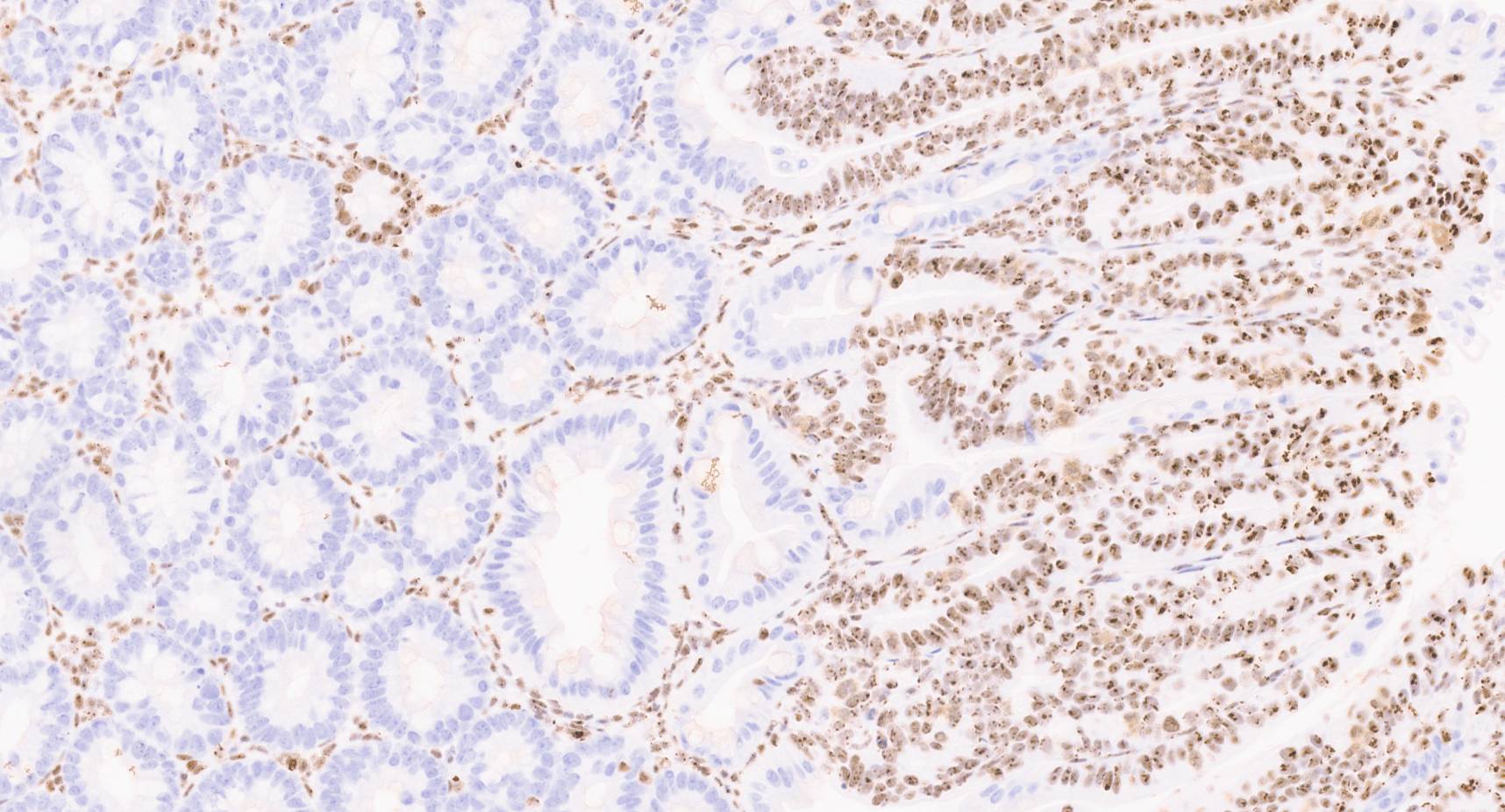
NPM1 is a well-established MYC target gene, already familiar to the cancer research community as the most frequently mutated gene in acute myeloid leukemia (AML). In fact, the World Health Organization recognises NPM1-mutated AML as a distinct entity among hematopoietic malignancies. Because of its prominence, there are ongoing efforts aimed at directly targeting NPM1. Interestingly, while NPM1 is rarely mutated in solid tumours, it is often highly overexpressed. We confirmed this pattern in our own analyses, observing significantly elevated expression of NPM1 across multiple human cancer types, with the highest levels seen in colorectal cancer.
The first surprise
Before diving into the cancer-promoting roles of NPM1, we first wanted to understand whether its loss is tolerated under normal conditions. This was one of our biggest concerns; tissue toxicity is a major hurdle when considering any potential drug target. NPM1 loss is known to be embryonic lethal, it is classified as an essential gene in many cancer cell lines, and most prior studies have examined its role in a heterozygous state, often within specific lineages such as the hematopoietic system. To our surprise, however, whole-body deletion of Npm1 was remarkably well tolerated long term in mice. Even more strikingly, when Npm1 was co-deleted with Apc, it significantly suppressed the hyperproliferation typically driven by high WNT signalling in both the intestine and liver.
In a mouse model of CRC, where tumour formation is driven by Apc loss, we observed that tumours could not form without expressing NPM1. Furthermore, Npm1 deletion not only reduced proliferation, but also significantly extended overall survival following additional oncogenic KRAS activation, a key event in CRC progression that transforms manageable adenomas into aggressive, drug-resistant cancers.
The second surprise
Naturally, our next step was to uncover the molecular mechanisms behind NPM1's striking effects on tumourigenesis. Surprisingly, RNA sequencing revealed no major transcriptional differences between tissues lacking APC alone and those with Apc and Npm1 co-deletion. We are cautious on a good day about relying too heavily on RNA levels - mRNA does not always reflect protein levels - but this result still caught us by surprise. So much so that another researcher in the team re-analysed the data from scratch only to confirm the result. Somehow, NPM1 loss was blocking the effects post-transcriptionally.
To get around this unexpected roadblock, we took two parallel approaches. The first was to investigate the p53 pathway, since early reports on Npm1 deletion had noted increased p53 activity. To test this hypothesis, we co-depleted p53 alongside NPM1 in vivo. And just as we began these experiments the COVID-19 pandemic hit. With major restrictions on animal work for over 18 months, our timelines suddenly got a lot more… interesting! The second approach was a broader exploration of post-transcriptional regulation: we turned to ribosome profiling (ribo-seq) to see whether specific mRNAs were being differentially translated, and paired this with proteomics to directly measure changes in protein abundance.
Don’t always believe the RNA!
Depleting p53 restored both hyperproliferation and tumourigenesis in the Apc-Npm1-deleted intestine, confirming that the p53 pathway plays a key role in the phenotypes triggered by NPM1 loss, even though typical transcriptional signatures of p53 activation were absent. What was even more intriguing was what we uncovered next. The ribosome profiling and proteomics data showed opposite enrichment patterns for key pathways: some pathways were positively enriched in the ribo-seq data but negatively enriched at the protein level. Ribo-seq also revealed that, in the absence of NPM1, more ribosomes were “stuck” at the beginning of mRNA coding sequences, pointing to a bottleneck in translation initiation. This led us to hypothesise a general translation stress response. And indeed, markers of protein synthesis stress were elevated upon NPM1 depletion. Strikingly, treating cells with inhibitors of this stress response not only rescued the hyperproliferation phenotype but also suppressed the induction of p53, suggesting a mechanistic link between translation stress and p53 activation.
Lessons learned and new avenues
This 7.5-year journey has been a roller-coaster of emotions and unexpected discoveries. Beyond reaffirming not to trust the mRNA, our work highlights NPM1 as a promising, if somewhat familiar, target for cancer therapy. Importantly, it suggests that efforts to target NPM1 in AML might also benefit patients with solid tumours. Our findings also strengthen the growing hypothesis that targeting translation could be a powerful cancer treatment strategy. Cancer cells depend on increased protein synthesis to sustain rapid growth, and while normal tissue homeostasis remains largely unaffected by NPM1 loss, hyperproliferative cells driven by oncogenic signalling do not. That said, we have only just begun to uncover how NPM1 influences translation and under what specific conditions. The only certainty is that working through social distancing restrictions was an unexpected experiment in itself, hopefully one that we will not have to repeat!
Follow the Topic
-
Nature Genetics
This journal publishes the very highest quality research in genetics, encompassing genetic and functional genomic studies on human and plant traits and on other model organisms.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in