CaTCH - A time machine to study clonal evolution
Published in Bioengineering & Biotechnology

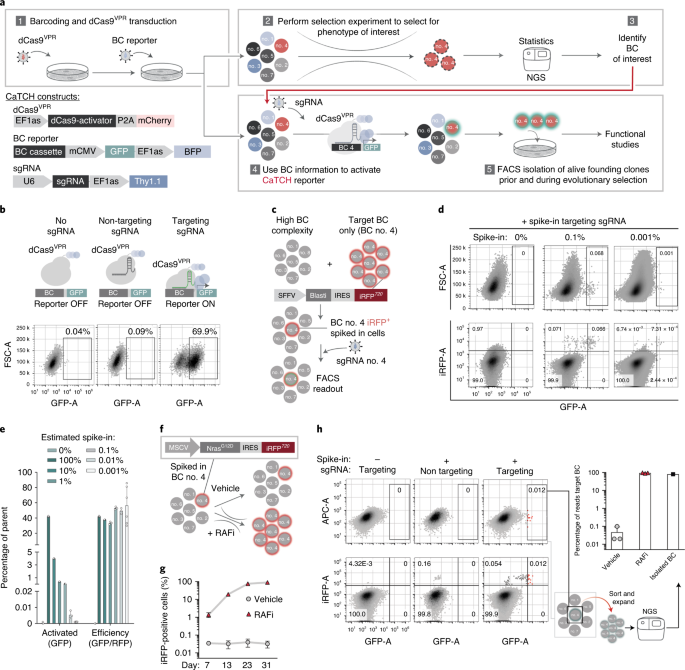
A main focus of the Obenauf lab at the IMP in Vienna is to gain insights into evolution of tumors upon treatment with targeted therapies, which inevitably fail due to the emergence of resistance. When I joined the lab, I became intrigued by the question on how targeted therapy resistance develops in a tumor. Is it simply a selection of fully resistant cell clones which pre-exist and initially reside within the tumor at a low abundance before they take over during treatment? Or are certain cells actively adapting to the therapy and become resistant only when the therapeutic pressure is applied? Many efforts had been undertaken to tackle this question, but conclusions were largely of indirect nature. This sparked the idea of isolating the same single cell clone before and after therapy, with the goal to directly characterize resistant clones but in their pre-treatment or treatment-naïve state. But how to identify these clones when we will only know which clones to look at after the selection had been applied? This is where we jokingly realized: “we needed a time machine”. However, we soon concluded that this could be possible by only fulfilling two prerequisites. First, we would need to keep track of the clonal identity within a large population, which can be easily done with genetic lineage tracing tools like DNA barcodes. Second, we would need to identify and isolate the clones of interest from the complex population. Here, CRISPR came into play. CRISPR would allow us to not only use DNA barcodes as unique identifiers but when optimized for sgRNA binding and coupled to a fluorescent reporter, they could also be used as molecular switches, which can be turned on by barcode specific sgRNAs. Thus, we could first barcode a heterogeneous cancer cell population, apply a targeted therapy to select for resistant clones and read-out their barcodes by next generation sequencing. In a second step we could use a copy of the starting population, which was initially maintained and never treated. By transducing this population with a sgRNA complementary to the previously identified resistant clone’s barcode, we could specifically activate the reporter allowing the isolation of desired clones, i.e. by fluorescence activated cell sorting. This would leave us with a clonal pair of cells, once in their treatment-naïve and once in their resistant state. The comparison of their drug responses can then give direct evidence for a pre-existing or an acquired resistance.
When it came to design the switch and decide on an activation strategy, we tried to be particularly careful. We knew that the desired clones might be of very low abundance, so we needed a highly complex barcoding library and a high sensitivity of the barcode-switch activation. We considered many ideas and CRISPR-tools and found CRISPRa-induced reporter activation to be the most promising approach. Using dCas9-VPR, we could pre-engineer the entire cell population to express this factor and would only need to deliver an easy to transduce sgRNA at the isolation step, which should strongly increase resolution. Further, we could generate a semi-randomly designed barcoding library offering several sgRNA targets per clone and a nearly limitless barcode complexity at a fraction of the costs of a fully designed library.
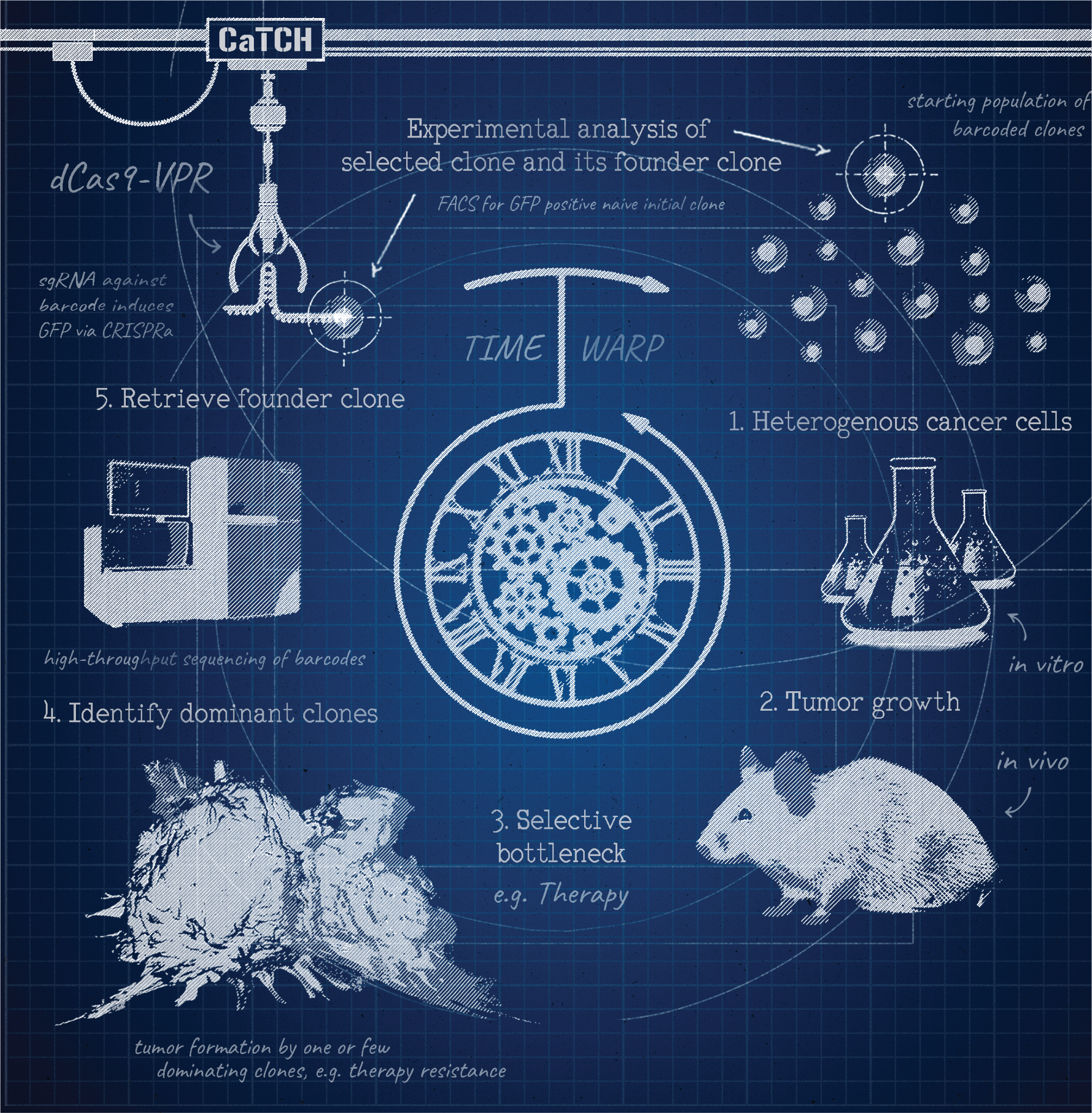 The workflow of CaTCH is visualized as a blueprint. 1. A heterogeneous cell population (of i.e. cancer cells) is barcoded with the CaTCH components. 2. The barcoded cells are cultured to establish a larger population for in vitro treatments or for injection based in vivo models. 3. A selective bottleneck (i.e. cancer therapy) is applied to select for a surviving cell population (i.e. of therapy resistant clones). 4. The dominant surviving clones are identified by their barcodes using Next Generation Sequencing. 5. Clones of interest can be isolated from the selected (step 3) and from the starting population (step 1). This is achieved by transduction of a sgRNA which activates the clones' CaTCH reporter in a barcode specific manner. Reporter activation allows isolation of these clones by FACS (indicated by the mechanical grabber). The selected and its founder clone can then be experimentally analyzed.
The workflow of CaTCH is visualized as a blueprint. 1. A heterogeneous cell population (of i.e. cancer cells) is barcoded with the CaTCH components. 2. The barcoded cells are cultured to establish a larger population for in vitro treatments or for injection based in vivo models. 3. A selective bottleneck (i.e. cancer therapy) is applied to select for a surviving cell population (i.e. of therapy resistant clones). 4. The dominant surviving clones are identified by their barcodes using Next Generation Sequencing. 5. Clones of interest can be isolated from the selected (step 3) and from the starting population (step 1). This is achieved by transduction of a sgRNA which activates the clones' CaTCH reporter in a barcode specific manner. Reporter activation allows isolation of these clones by FACS (indicated by the mechanical grabber). The selected and its founder clone can then be experimentally analyzed.
The system, which we termed CaTCH (CRIPSRa tracing of clones in heterogeneous cell populations), performed very well in carefully controlled tests. Thus, we soon proceeded to challenge its applicability in a resistance selection experiment in vivo to tackle our initial question of how resistance develops. To our surprise we found that even though resistant tumors consisted of only a handful of clones (from initially tens of thousands!), none of the treatment-naïve founder clones was fully resistant and seemingly all clones within the treatment-naïve starting population had the potential to become resistant. We further could attribute this to the de novo generation of resistance-causing mutations in certain clones, challenging the common notion of pre-existing resistance-mutations. We are very curious to follow up on these findings in future studies.
The benefit of having developed a general biotechnological tool for clonal isolations is that it is not only limited to the application, which we had intended to use it for. The isolation and characterization of founding clones holds unique potential for understanding physiological and disease processes from differentiation, over disease modeling, to regenerative medicine and can help to uncover novel biomarkers and therapeutic concepts. Therefore, we hope that our development will be accepted well by the research community and we are very excited about future applications of CaTCH.
Our corresponding paper has been published in Nature Biotechnology and can be found here: https://www.nature.com/articles/s41587-020-0614-0
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in