Cholinergic T cells at the center of neuro-immuno-oncology
Published in Cancer

Researchers have begun to recognize that, within the tumor microenvironment, interactions occur among nerve endings, local immune cells, and cancer cells that have a profound influence on tumorigenesis. Most primary tumor sites are innervated by nerves of the sympathetic and parasympathetic nervous systems. Sympathetic nerve “inputs”, relying on the neurotransmitter norepinephrine, tend to have tumor-promoting effects in various tissues. In contrast, parasympathetic nerves, which rely on the neurotransmitter acetylcholine (ACh), have effects on cancers that vary by tissue. For instance, surgical denervation of cholinergic inputs reduces the incidence of gastric malignancies but promotes pancreatic cancers. Determining the precise roles of cholinergic inputs in different cancers therefore requires much further effort.
Although ACh was initially isolated from the spleen back in 1929, its contribution to immune regulation has only recently been appreciated. Subsets of both T and B lymphocytes can express choline acetyltransferase (ChAT), the enzyme producing ACh, but how these cholinergic immune cells affect tumorigenesis is largely unknown. This paper highlights our lab’s interest in the roles of these cells in hepatocellular carcinoma (HCC). Both human and mouse livers are innervated only by sympathetic nerves, meaning that there is essentially no cholinergic nerve input in this organ. We wondered if there were any cholinergic immune cells in the liver and, if so, did they have a prominent function here? To answer this question, we devised a novel genetic model of HCC development in mice. As had already been reported, we observed that neither healthy nor HCC-bearing mouse livers harbored cholinergic nerves. However, we did find a substantial number of ChAT-expressing lymphocytes in livers, especially in those bearing HCC nodules.
HCC tumor antigens induce ChAT-expressing T cells
We analyzed these immune cells using our HCC model and detected gradual accumulations of CD4+ T cells and CD8+ T cells during HCC development. Indeed, the number of ChAT-GFP+ CD4+ T cells in HCCs significantly increased during tumor progression. We carried out single-cell RNA sequencing analyses of hepatic T lymphocytes and noted a marked shift from naïve T cells to effector T cells, particularly the induction of the Cxcr6+Pdcd1+ cell cluster, when HCCs were present. We also observed the induction of a range of ChAT-expressing T cells, including regulatory T cells (Tregs), PD-1+ conventional T cells (Tconvs), and proliferating T cells, in not only mouse HCCs but also in human HCC samples.
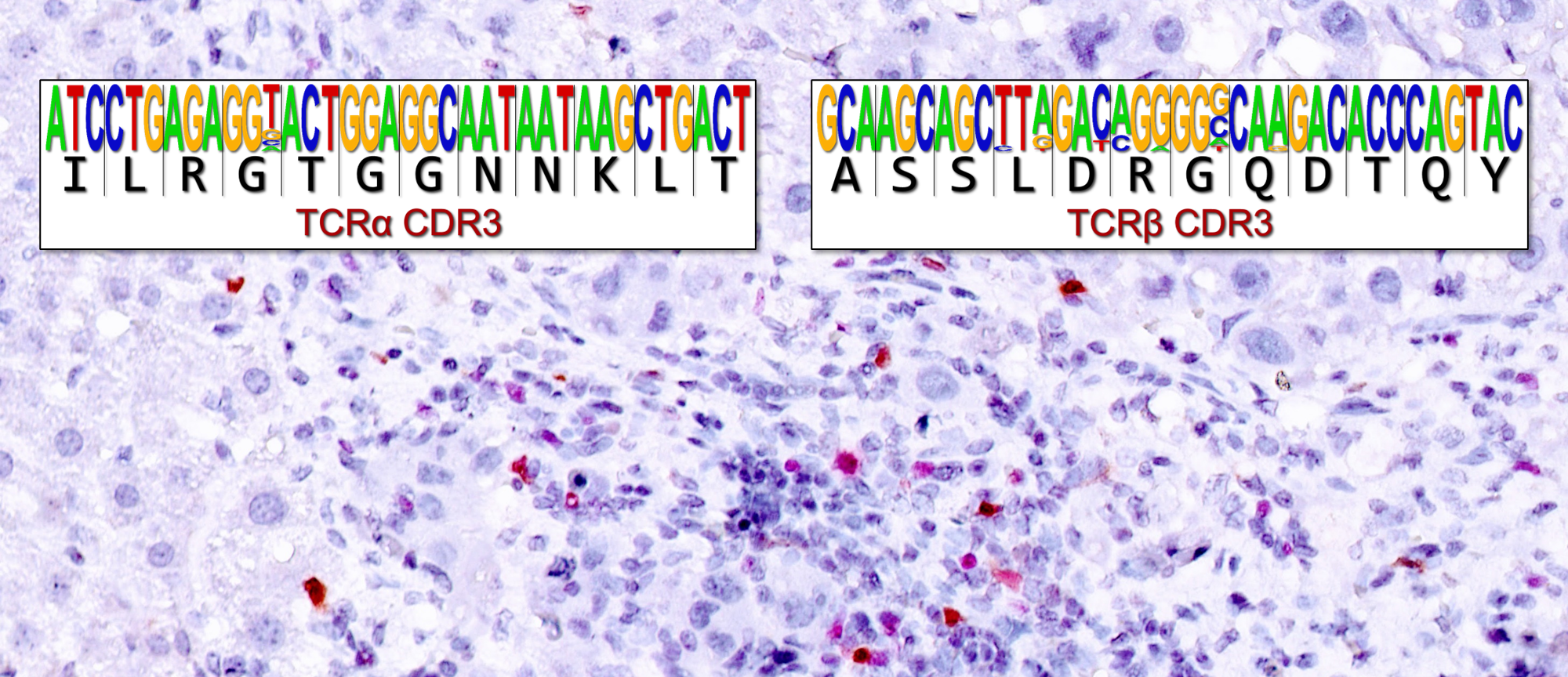
We next turned to single-cell TCR analysis and showed that the expansion of ChAT-expressing CD4+ T cells in mouse HCCs was TCR-specific and, unexpectedly, also TCR- convergent; that is, the most dominant TCR protein was encoded by 25 mRNA clonotypes. These clonotypes consisted of synonymous mRNA variants that used the same combination of V, D, and J genes but displayed distinct junctions of these segments in different HCC-bearing mice. As a result, the amino acid sequences of the TCR proteins expressed were identical due to codon degeneracy. Intriguingly, most of these clonotypes were predominantly observed in ChAT-GFP+ cells. We concluded that this TCR convergence across HCC-bearing mice meant that TCRs specific for HCC antigens had elicited the expansion of CD4+ T cells, particularly within the ChAT-GFP+CD4+ T cell compartment. We then devised an HCC model in which we could trigger the expression of a defined tumor antigen, and demonstrated that such an antigen could indeed induce ChAT-expressing T cells in liver cancer.
What are ChAT-expressing T cells doing during HCC development?
To answer this question, we crossed Chatflox mice with Cd4-cre mice so as to generate Chatfl/fl;Cd4-cre animals in which ChAT was genetically deleted specifically in T cells. We found that Chatfl/fl;Cd4-cre mice developed liver cancer much faster than their control littermates, with greater numbers of tumor nodules and heavier liver weights. In ChAT-competent control mice subjected to HCC induction, we observed immune cells clustered around MYC-expressing preneoplastic cells. These clusters were reduced in frequency and size in Chatfl/fl;Cd4-cre mice, suggesting that a lack of ChAT in T cells compromised the immunosurveillance of incipient liver cancer cells. Consistent with this finding, the infiltration of T cells into HCC-bearing livers was significantly decreased in Chatfl/fl;Cd4-cre mice. These results prompted us to hypothesize that ChAT might have cell-autonomous functions in T cells responding to HCC initiation.
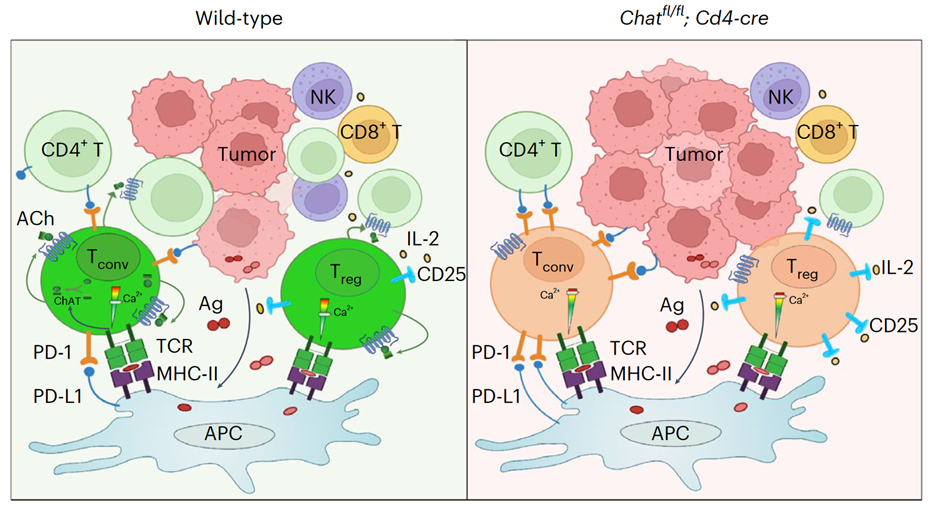
Acetylcholine binds to two types of receptors, namely muscarinic receptors and nicotinic receptors. The muscarinic receptor subtypes M1, M3, and M5 couple to the Gq/11 class of G proteins and trigger calcium signaling, whereas the M2 and M4 subtypes are generally linked with the Gi/o class of G proteins to inhibit cAMP synthesis. The nicotinic receptors are ACh-gated ionic channels. We found that the T cells in mouse HCCs expressed both muscarinic and nicotinic receptors, raising the possibility of an autocrine/paracrine loop of ACh signaling. Compared to control T cells, ChAT-deficient T cells exhibited a stronger calcium influx in response to TCR stimulation. Moreover, pre-stimulation with ACh prevented the nuclear translocation of NFAT triggered by TCR activation, and this effect depended on a muscarinic receptor. Therefore, we concluded that the cholinergic activity in T cells had suppressed TCR-induced Ca2+/NFAT signaling. However, this increased TCR activity in ChAT-deficient T cells contradicted the reduced T cell number observed in HCCs of Chatfl/fl;Cd4-cre mice, prompting us to investigate the cause of this discrepancy.
Why does the loss of ACh-regulated Ca2+/NFAT signaling promote HCC?
In general, the liver favors tolerance over immune responses, and HCCs provide a tolerogenic environment for T cells. Accordingly, we observed expanded Tregs and dysfunctional Tconvs in our mouse HCCs. When we used OVA to mimic a tumor antigen, the induced antigen-specific T cells were mainly Tregs and PD-1+ Tconvs, and ChAT was exclusively expressed by these two subsets. We then postulated that perhaps ACh was intrinsically regulating these two T cell subsets. Indeed, the Foxp3+ Tregs in HCCs of Chatfl/fl;Cd4-cre mice expressed substantially more CD25 than those in ChAT-expressing controls. Similarly, PD-1 expression was higher on Tconvs isolated from HCCs of Chatfl/fl;Cd4-cre mice. It is well known that expression levels of both CD25 and PD-1 in T cells are directly regulated by the Ca2+/NFAT pathway, and so these data were in line with the elevated TCR-induced calcium signaling in ChAT-deficient T cells. When we depleted CD25+ Tregs in Chatfl/fl;Cd4-cre mice or applied PD-1 blockade, HCC development was significantly reduced. We concluded that an absence of cholinergic signaling in T cells unleashes a hyperactivation of Treg cells and exacerbates Tconv dysfunction such that the anti-tumor immune response is impaired, allowing HCC progression.
With respect to the human situation, we perused public databases to confirm that T cells in human liver cancers also express cholinergic receptors. Moreover, high expression of the CHRM3 or CHRM5 genes encoding Ca2+ signaling-related muscarinic receptors in HCC patient samples correlated positively with a favorable prognosis. We concluded that a T cell cholinergic program likely also plays a protective role against liver cancer development in humans, a possibility we continue to explore.
Collectively, the data presented in our paper support a model (see Figure) in which tumor antigens induce the expansion of ChAT-expressing Tregs and PD-1+ Tconvs. ACh produced by these T cells acts in an autocrine/paracrine manner to modulate TCR-induced Ca2+ signaling to prevent overactivation of this pathway (left panel). In the absence of such cholinergic modulation (right panel), the Ca2+/NFAT pathway becomes hyperactivated, leading to increased immunosuppression by Tregs and aggravating the dysfunction of Tconvs. The result is compromised anti-HCC immunity.
Follow the Topic
-
Nature Cancer
This journal aims to provide a unique forum through which the cancer community will learn about the latest, most significant cancer-related advances across the life, physical, applied and social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cancer Neuroscience: from mechanisms to therapy
Publishing Model: Hybrid
Deadline: Jan 30, 2027


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in