Controlling Oncogenic KRAS Signaling Pathways with a Palladium-Responsive Peptide
Published in Chemistry
In the spring of 1948, while playing with a strip of paper in which he had drawn a polypeptide chain, Linus Pauling built the first model of an α-helix. This initial insight on the three-dimensional arrangement of proteins led him, together with Robert B. Corey and Herman R. Branson, to a foundational publication in structural biology, some say as important as that by Watson and Crick describing the DNA duplex, in which they predicted the atomic structure of the α-helix (Pauling et al., 1951). Over the years, researchers have realized that α-helices are not only one of the fundamental protein building blocks but also frequent mediators in protein-protein and protein-DNA interactions that regulate key signaling pathways. It has been estimated that roughly 60% of all protein complexes feature α-helical interfaces (Jochim and Arora, 2010). α-helices are, thus, a key ingredient of the language that proteins use to communicate, and so they have become a hot target for the development of new protein-protein interaction inhibitors. But developing such α-helical inhibitors is not straightforward because α-helices are delicate structures and short peptide sequences do not usually maintain their helical structure when extracted from their parent protein. This can be understood going back to Pauling’s folded paper model, which you would need to hold together with your hands or a piece of tape, otherwise, the helix unrolls and the peptide is straightened out. Much like it, at the molecular level, α-helices are, at best, marginally stable structures because the energy required to fold them into the correct helical arrangement, what is known as the entropic cost, is hardly compensated by the little energy gained upon folding, the enthalpic term. Unfolded helices are pretty much useless: they do not present their functional groups in the right place to create a binding site, and the energy required to fold them, taxes the formation of their complexes so much that the binding affinity would be too low.

One of the most successful strategies to develop α-helical inhibitors has been the preorganization of the α-helical conformation to reduce the entropic term. This can be done by introducing crosslinks between side chains in the same face of the α-helix, which are typically known as (i,i+4) and (i,i+7) positions according to the number of amino acid residues between the crosslink (de Araujo et al., 2014; Lau et al., 2015). Examples of this strategy include lactam bridges between the side chains of Lys and Asp residues,(Hoang et al., 2019; Shepherd et al., 2005) salt bridges between charged Glu and Lys residues (Marqusee and Baldwin, 1987), and metal ion coordination with appropriately installed His residues in adjacent helical turns (Ghadiri and Choi, 1990; Ghadiri and Fernholz, 1990; Kelso et al., 2000; Ma et al., 2009). Currently, most crosslinked peptidomimetics rely on hydrocarbon linkers formed by ruthenium-catalyzed ring-closing metathesis (RCM) reactions between unnatural alkenyl amino acids (Cromm et al., 2015). In contrast to these constitutively active inhibitors, locked into stable α-helical conformations by permanent clamps, stimuli-responsive peptides that change their affinity in response to an external input could offer new opportunities in basic research, as well as for spatiotemporal controlled therapeutic applications. Thus, going beyond the classic metal-stabilized α-helices, and considering our previous studies in the field of DNA binding (Gómez-González et al., 2021; Learte-Aymamí et al., 2020a; Rodríguez et al., 2016a, 2016b), we hypothesized that metal chelation could be applied for the development of switchable α-helical peptides that modify their folding and binding affinity in response to an external stimulus, namely, the presence of a Pd(II).
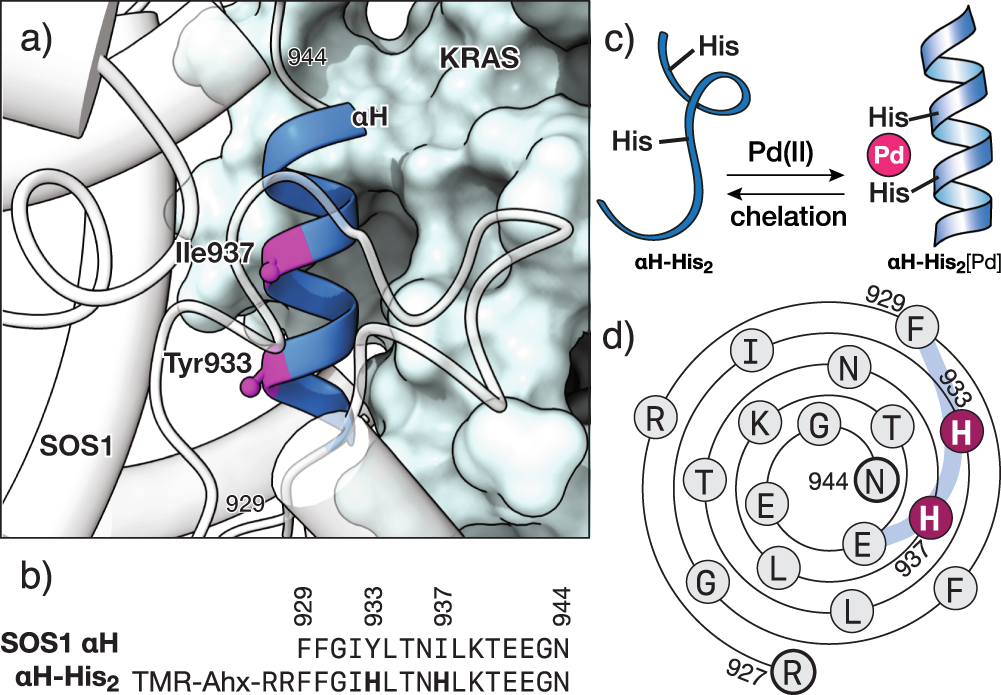
Our design takes as a starting point the structure of the KRAS/SOS1 complex, from which we isolated the αH helix of SOS1 bound to KRAS. In this hypothetical complex, we observed that Tyr933 and Ile937 would be located in the solvent-exposed face of the αH helix fragment and distanced so that their side chains, when replaced by His residues, would be in register and appropriately oriented to chelate a Pd(II) center. Thus, the peptide αH-His2, engineered to include two (i,i+4) His residues in a short fragment of the SOS1 αH helix, binds KRAS only after the addition of a Pd(II) source and subsequent chelation.

Fig. 1. Design of the metal-responsive KRAS peptide inhibitor. Top: Detail of the αH helix of SOS1 complexed with RAS (PDB code 1BKD) indicating the residues Tyr933 and Ile937 that are replaced by the metal chelating His residues; the structure of SOS1 is shown in white. Bottom: Sequence of the natural αH helix of SOS1 and the mutant peptide αH-His2 used in this study, highlighting the two engineered His residues.
More importantly, we also show that such coordination is reversible so that the metallopeptide αH-His2[Pd] can be disassembled by the addition of an external Pd(II) chelator. This coordination-induced folding/unfolding can be easily monitored by Circular Dichroism spectroscopy, which shows the typical α-helix signature only in the presence of the metal ion (see Figure below) (Learte-Aymamí et al., 2017, 2020b).

Fig. 2. Circular Dichroism studies show that the α-helical content of the peptide αH-His2 can be switched. Circular dichroism of a 20 μM solution of αH-His2 (dashed line) and the spectrum 30 min after addition of 1 eq. of cis-PdCl2(en) showing the signature of an α-helix. The addition of the Pd(II) chelator DEDTC disassembles the helix, which results in a reduction of the intensity of the CD spectrum at 208 and 222 nm.
In addition to demonstrating the possibility of controlling the conformation of the bioactive αH-His2 peptide through metal chelation, in this proof-of-concept work, we challenge the current paradigm for the design of α-helix peptidomimetics, which seeks to maximize the α-helical content to avoid the entropic penalty associated with the folding of the peptide into its bioactive helical conformation. We demonstrate that such drastic preorganization is not required and that marginally stable helices can act as effective inhibitors through a folding-upon-binding mechanism, just like the natural Intrinsically Disordered Proteins that exploit this process to modulate protein activity. Thus, despite the modest effect of Pd(II) chelation in the overall helical content of the αH‑His2 peptide, Pd(II) coordination led to a dramatic change in the affinity for KRAS, so that while no significant binding was observed by fluorescence anisotropy when the free peptide αH‑His2 was incubated with KRAS, titrations with the metallopeptide αH‑His2[Pd] resulted in clear binding isotherms with tight dissociation constants of 345 nM and 294 nM for KRASG12C and KRASG12V, respectively. This effect is consistent with an induced folding-upon-binding mechanism facilitated by the nucleation of the bioactive α helical conformation when the Pd(II) coordinates the two His residues. Therefore, although the coordination of the metal ion does not provide enough energy to fold the metallopeptide αH‑His2[Pd] into an α-helix by itself, it is enough to do so in the presence of KRAS and the additional interactions with the protein that stabilize the bound helix. This behavior reproduces that of Intrinsically Disordered Proteins (IDPs) or regions of proteins (IDRs), which are typically involved in protein-protein interactions regulating signaling pathways, that rely on their transition from a disordered to a folded state upon recognition of their physiological partners (Toto et al., 2020). This mechanism allows high specificity, ultra-sensitivity, and switching behavior (Teilum et al., 2021), which might also be desirable properties in designed peptide inhibitors.
Finally, the preformed metallopeptide αH‑His2[Pd] was internalized by A549 cells, as demonstrated by fluorescence microscopy, and the peptide inhibited the MAPK RAF-MEK-ERK cascade in a dose-responsive manner, as demonstrated by the measured decrease in the levels of phosphorylated ERK. To our knowledge, this is the first demonstration of a designed metallopeptide that can modulate a signaling pathway in living cells. Our approach uncovers new possibilities for the design of dynamic α-helix peptidomimetics that exploit the folding-upon-binding mechanism and can be extended to other biological targets.
References
de Araujo, A.D., Hoang, H.N., Kok, W.M., Diness, F., Gupta, P., Hill, T.A., Driver, R.W., Price, D.A., Liras, S., and Fairlie, D.P. (2014). Comparative α-helicity of cyclic pentapeptides in water. Angew. Chem. Int. Ed. 53, 6965–6969.
Cromm, P.M., Spiegel, J., and Grossmann, T.N. (2015). Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 10, 1362–1375.
Ghadiri, M.R., and Choi, C. (1990). Secondary structure nucleation in peptides. Transition metal ion stabilized α-helices. J. Am. Chem. Soc. 112, 1630–1632.
Ghadiri, M.R., and Fernholz, A.K. (1990). Peptide architecture. Design of stable α-helical metallopeptides via a novel exchange-inert ruthenium(III) complex. J. Am. Chem. Soc. 112, 9633–9635.
Gómez-González, J., Pérez, Y., Sciortino, G., Roldan-Martín, L., Martínez-Costas, J., Maréchal, J.-D., Alfonso, I., Vázquez López, M., and Vázquez, M.E. (2021). Dynamic Stereoselection of Peptide Helicates and Their Selective Labeling of DNA Replication Foci in Cells. Angew. Chem. Int. Ed. 60, 8859–8866.
Hoang, H.N., Wu, C., Hill, T.A., Dantas de Araujo, A., Bernhardt, P.V., Liu, L., and Fairlie, D.P. (2019). A Novel Long-Range n to π* Interaction Secures the Smallest known α-Helix in Water. Angew. Chem. Int. Ed. 58, 18873–18877.
Jochim, A.L., and Arora, P.S. (2010). Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem. Biol. 5, 919–923.
Kelso, M.J., Hoang, H.N., Appleton, T.G., and Fairlie, D.P. (2000). The First Solution Structure of a Single α-Helical Turn. A Pentapeptide α-Helix Stabilized by a Metal Clip. J. Am. Chem. Soc. 122, 10488–10489.
Lau, Y.H., de Andrade, P., Wu, Y., and Spring, D.R. (2015). Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 44, 91–102.
Learte-Aymamí, S., Curado, N., Rodríguez, J., Vázquez, M.E., and Mascareñas, J.L. (2017). Metal-Dependent DNA Recognition and Cell Internalization of Designed, Basic Peptides. J. Am. Chem. Soc. 139, 16188–16193.
Learte-Aymamí, S., Rodríguez, J., Vázquez, M.E., and Mascareñas, J.L. (2020a). Assembly of a Ternary Metallopeptide Complex at Specific DNA Sites Mediated by an AT-Hook Adaptor. Chem. Eur. J. 26, 8875–8878.
Learte-Aymamí, S., Vidal, C., Gutiérrez-González, A., and Mascareñas, J.L. (2020b). Intracellular Reactions Promoted by Bis(histidine) Miniproteins Stapled Using Palladium(II) Complexes. Angew. Chem. Int. Ed Engl. 59, 9149–9154.
Ma, M.T., Hoang, H.N., Scully, C.C.G., Appleton, T.G., and Fairlie, D.P. (2009). Metal clips that induce unstructured pentapeptides to be α-helical in water. J. Am. Chem. Soc. 131, 4505–4512.
Marqusee, S., and Baldwin, R.L. (1987). Helix stabilization by Glu-...Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. USA 84, 8898–8902.
Pauling, L., Corey, R.B., and Branson, H.R. (1951). The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 37, 205–211.
Rodríguez, J., Mosquera, J., Vázquez, M.E., and Mascareñas, J.L. (2016a). Nickel-Promoted Recognition of Long DNA Sites by Designed Peptide Derivatives. Chem. Eur. J. 22, 13474–13477.
Rodríguez, J., Mosquera, J., García-Fandiño, R., Vázquez, M.E., and Mascareñas, J.L. (2016b). A designed DNA binding motif that recognizes extended sites and spans two adjacent major grooves. Chem. Sci. 7, 3298–3303.
Shepherd, N.E., Hoang, H.N., Abbenante, G., and Fairlie, D.P. (2005). Single turn peptide α helices with exceptional stability in water. J. Am. Chem. Soc. 127, 2974–2983.
Teilum, K., Olsen, J.G., and Kragelund, B.B. (2021). On the specificity of protein-protein interactions in the context of disorder. Biochem. J. 478, 2035–2050.
Toto, A., Malagrinò, F., Visconti, L., Troilo, F., Pagano, L., Brunori, M., Jemth, P., and Gianni, S. (2020). Templated folding of intrinsically disordered proteins. J. Biol. Chem. 295, 6586–6593.
I graduated in Chemistry from the Universidade de Santiago de Compostela in 1996 (with honors), and worked on my PhD from 1996 to 2001 under the supervision of Prof. José Luis Mascareñas developing new DNA-binding peptides. In 2001 I received the Human Frontier Science Program long-term fellowship and joined the group of Prof. Barbara Imperiali at the Massachusetts Institute of Technology, where I worked for three years, from 2001 to 2004 on the development of caged compounds and fluorescent probes as tools to understand complex phosphorylation pathways involved in cell motility.
I returned to Santiago with a Ramón y Cajal contract in 2004 and was habilitated in 2007. Since 2010 I am enjoying an Associate Professor position at the Organic Chemistry Department, and in 2011 I became a member of the Center for Research in Biological Chemistry and Molecular Materials (CiQUS). I was Habilitated for full Professor in 2020.
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in