Creating the next generation of protein sequence reference libraries for marine microbial eukaryotes
Published in Ecology & Evolution, Microbiology, and Protocols & Methods
Covering 70% of the Earth's surface, our oceans are teeming with life. Each droplet of seawater is a microcosm, home to millions of single-celled organisms including bacteria and the more complex eukaryotes. Essential to marine ecosystems, eukaryotic phytoplankton and cyanobacteria form the basis of food webs, converting solar energy into biomass in the photic zone. This primary production not only fuels the microbial ecosystem but also plays a crucial role in the global cycling of vital elements like carbon and nitrogen, ultimately supporting fisheries that are key to human nutrition. The advent of environmental gene sequencing technologies in the early 2000s marked a turning point in marine microbiome research, offering an unprecedented window into these in situ communities, many of which had previously evaded isolation and study in laboratory settings.
To decode the genetic mysteries of marine microbiomes, researchers employ two main techniques: 'metagenomics' and 'metatranscriptomics'. Metagenomics focuses on the genomic DNA sequences of microbial cells, providing a snapshot of the potential genetic capabilities within a community. Metatranscriptomics, on the other hand, targets messenger RNA transcripts, revealing which genes are actively being expressed at any given moment. Both methods generate millions of nucleotide sequences. To make sense of these sequences, a process called taxonomic annotation is employed, where each sequence is compared to known reference sequences from established taxa. This comparison is crucial for understanding the genetic diversity and functional potential of marine microbial communities. Bacterial genomes, known for their compactness and relative simplicity, are well-suited for sequencing, leading to thousands of bacteria with fully sequenced reference genomes. Eukaryotes, in contrast, present a more formidable challenge. Their genomes are not only larger—typically by an order of magnitude—but also more intricate, with a complex mix of repetitive elements and spliced transcripts. Despite these hurdles, eukaryotic studies benefit from a unique feature: the poly-adenylation tags on messenger RNA transcripts. This allows for a selective focus on the active, protein-coding parts of the genome, simplifying the task of sequencing and understanding eukaryotic genetic material.
A major milestone in expanding marine eukaryote reference sequences was achieved with the 2014 launch of the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP). This collaborative initiative sequenced the RNA of nearly seven hundred marine microbial eukaryotes, many of which were previously unrepresented in terms of their genetic makeup. This effort significantly increased the number of reference taxa, from a mere handful to hundreds, thereby enhancing research capabilities in identifying environmental metatranscriptome sequences. A decade later, MMETSP remains a cornerstone in most marine eukaryotic reference sequence libraries. However, despite this progress, the number of marine microbial eukaryotes with fully sequenced genomes or transcriptomes is still dwarfed by the approximately 200,000 taxa identified through 18S ribosomal RNA in the PR2 rRNA metabarcode reference database. Nevertheless, the pool of sequenced reference taxa continues to expand, enriched by both large-scale and smaller, focused sequencing projects, gradually filling in the vast genetic landscape of marine microbial eukaryotes.
In response to the increasing array of new reference sequences, several databases have been developed to facilitate marine eukaryotic metatranscriptome annotation. These databases incorporate not only the foundational MMETSP transcriptomes but extend to include hundreds of other genomes and transcriptomes. However, despite their utility, practical use of these libraries face significant challenges. One primary concern is ensuring that they remain up-to-date while catering to diverse scientific needs. Current libraries draw from varied data sources, leading to inconsistencies in coverage and content. Moreover, they often overlook the contributions from smaller-scale sequencing projects, which collectively add considerable diversity. To address specific research queries, many researchers have resorted to creating 'custom reference sequence libraries', supplementing existing ones with additional data. This practice, while flexible, can hinder reproducibility due to the lack of standardized documentation for these customized resources.
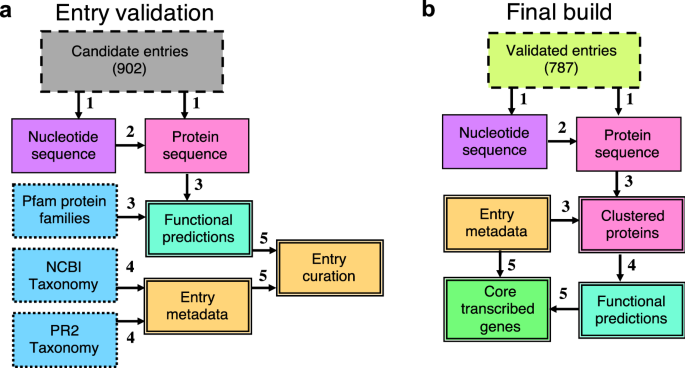
In the Armbrust lab, our extensive work with marine eukaryotic metatranscriptomes, derived from numerous field cruises, has heavily relied on reference sequence libraries for identifying environmental gene sequences. Our research, focusing on key phytoplankton genera and the nuanced differences between closely related genes, underscores the need for the most precise annotations we can achieve. Recognizing gaps in existing libraries, we incorporated new, publicly available reference taxa and identified the necessity for streamlined, well-documented releases of custom reference sequence libraries. To address these needs, we have created the Marine Functional EukaRyotic Reference Taxa (MarFERReT) library, a sequence library tailored for eukaryotic metatranscriptome annotation (Figure 1). Grounded in a flexible, open-source framework, MarFERRet version 1.1 aggregates over 900 genome and transcriptome data sets from diverse sources, including MMETSP re-assemblies and several large data repositories, as well as smaller-scale studies. This library represents a significant step forward in compiling comprehensive and diverse genetic information for marine eukaryotes.

Recognizing the importance of data integrity, particularly against sequence cross-contamination, we incorporated quality control measures into MarFERRet's workflow. Each of the 900 data sets underwent technical validation steps, including an estimate of potential cross-contamination inferred from ribosomal protein analysis. After this thorough evaluation, we selected 800 high-quality genomes and transcriptomes for the final build of our reference library, ensuring a reliable resource for accurate taxonomic inference of environmental sequences. Detailed in the MarFERReT v1.1 publication, this comprehensive resource is now available in a Zenodo repository for immediate access and utilization. The build process, from ingestion of downloaded raw data to the final protein sequence products, is encapsulated in a containerized workflow on our code repository. This not only ensures reproducibility of the current build but also facilitates the development of derivative databases, tailored with new or modified datasets.
Protein sequence libraries have served as the backbone of environmental sequence annotation, offering deep insights into the dynamic genetic activities of in situ microbial communities. Beyond direct annotation, protein libraries are now extending their utility as foundational data sets in an developing field: protein language models (pLMs). Analogous to text-based large language models like GPT-3.5, pLMs are trained on vast arrays of protein sequences, opening new frontiers in biological research. As this field matures, the role of comprehensive and carefully curated protein datasets, such as those in MarFERReT, may become increasingly crucial for downstream model fine-tuning. As the development of pLMs finds its footing, there is the potential to propel advancements in AI-driven exploration of large complex environmental data sets.
We believe MarFERReT v1.1 stands as a valuable tool in its current form, yet we have designed it with ongoing growth and refinement in mind. We have plans for the development of MarFERReT version 1.2, slated for an early 2024 release, which will introduce dozens of additional reference taxa. Embracing the ethos of open-source development, we invite the scientific community to engage with MarFERReT, enhancing its codebase, content, and documentation. Recognizing the continued need for customized libraries, we hope MarFERReT will serve as model for flexible documentation and reproducibility and empower exciting new discoveries in the molecular ecology of the oceans.
Follow the Topic
-
Scientific Data
A peer-reviewed, open-access journal for descriptions of datasets, and research that advances the sharing and reuse of scientific data.

Related Collections
With Collections, you can get published faster and increase your visibility.
Computer vision in plant science and agriculture
Publishing Model: Open Access
Deadline: Oct 10, 2026
Wearable and Computer Vision Data for Health and Behaviour Research
Publishing Model: Open Access
Deadline: Aug 08, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in