CRISPR Off-Targets: One SNP can make all the difference
Published in Bioengineering & Biotechnology, Protocols & Methods, and Cell & Molecular Biology

In 2018, Nature Medicine published back-to-back manuscripts that claimed CRISPR-based genome editing could enrich for cancer mutations1,2. While the articles themselves were not exactly alarmist, the headlines they garnered certainly were (see below). This apparent cancer scare thus sent share prices of CRISPR companies into freefall as investors reconsidered the toxicity potential of this powerful technology.
Because the uniting focus of Matthew Porteus’ lab at Stanford is CRISPR-based genome editing for therapeutic applications, we were quite alarmed by these toxicity claims. Right after the release of these manuscripts, we scheduled an “emergency” journal club to become informed on this emerging topic. As we read the papers in depth, we became less concerned. CRISPR didn’t cause cancer mutations, instead the authors spiked cancer cells into the pool of cells being edited and found that (big surprise) cancer cells were pretty robust and tolerated CRISPR-mediated DNA breaks better than normal cells. Therefore, CRISPR genome editing enriched for cancer mutations if, and only if, they were already present at the time of editing. Because virtually all therapeutic editing occurs in patients without cancer, we felt this fact alone made these findings overblown and unfortunately alarmist.
Of course, as a genome editing lab, we wanted the data to be faulty or untrue. But good scientists correct for their biases and listen to data. So we decided to generate our own. To do so, we partnered with sequencing giant Illumina to perform ultra-deep sequencing of over 500 cancer genes following CRISPR genome editing of primary human cells. This set of genes represents an unbiased set of cancer-associated genes (the set of genes that would have Tier 1 level of concern if the process did create mutations in them). We then applied a clinical diagnostics sequencing kit on a clinical genome editing workflow. We used the same sequencing panel that pathologists use to detect oncogenes in patients with cancer. We evaluated a genome editing model used clinically, ex vivo culture and Cas9 delivery to primary (not cell line-derived) human hematopoietic stem and progenitor cells (HSPCs).
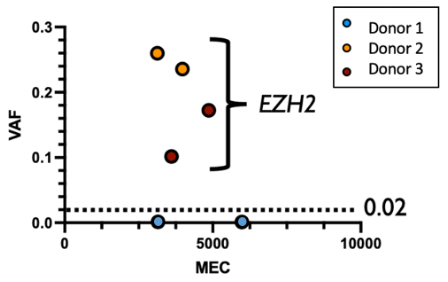
I recall our first meeting with the Illumina scientific team when we decided which guide RNAs (gRNAs) to use and—based on prior experience that demonstrated the exceedingly rare nature of unintended Cas9 off-target activity—I insisted we create an intentionally bad (i.e. non-specific) gRNA to ensure we find at least some unintended mutations. This gRNA was a near perfect match to a known oncogene, EZH2, and was meant to serve as a positive control.
In hindsight, we were so glad we included this bad guide, since those were the only mutations we found in any of the 500 cancer genes in any sample (see below)! To our astonishment, one of the HSPC donors had no detectable mutations in EZH2. Upon closer investigation, we found that this donor had a single pre-existing variant at the intended cut site. This alone was able to completely prevent Cas9 activity - even when we were trying to cause it. This accidental finding was a lucky albeit profound demonstration of the incredible specificity of HiFi Cas9.
During the revision process our results were further strengthened by reviewer suggestions. Their comments led us to expand the scope of sequencing to include the entire genome. Again, we found no detectable mutations introduced by Cas9 exposure other than at the intended cut site. Our methodology may have been unable to detect more rare consequences of Cas9 activity that have been reported, such as large-scale genomic rearrangements where long read sequencing may further map genomes without the need for informatic assembly. This will be an important area of consideration for future studies, which will help ensure that the safety of these editing tools keeps pace with the efficacy they are currently demonstrating in the clinic. It remains equally important to consider how preclinical experimental approaches—such as p53 mutant spike-in (compared to using populations with no pre-existing p53 mutations), genomic instability of cell lines (compared to genomically stable primary cells), stable and persistent Cas9 expression (compared to transient delivery of a high fidelity Cas9), use of research reagents that create cellular stress (compared to reagents designed to minimize cellular stress), etc.—can fail to capture meaningful and deliberate nuance of clinical implementation efforts.
Overall, we are proud of this data-based exploration of a topic with major translational ramifications. Patients are in need of curative therapies and our responsibility remains to introduce them safely. Our study further demonstrates how academia and industry collaborations can work together to serve the broader scientific and clinical community. As a genome editing lab, we were delighted that the data we generated reinforced the apparent safety of these genome editing tools. However, if we had found the opposite—that CRISPR did introduce or enrich for cancer mutations—we would have immediately published this result to alert the broader community. We knew our work was important as the findings, positive or negative, would yield progress for the genome editing field and, more fundamentally, for patients.
Citations:
1. Ihry, R.J. et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 24, 939-946 (2018).
2. Haapaniemi, E., Botla, S., Persson, J., Schmierer, B. & Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 24, 927-930 (2018).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in