Cross-catenation between position-isomeric metallacages
Published in Chemistry
Chemists introduced supramolecular interaction binding sites into ligand’ framework, combined with metal acceptors to synthesis supramolecular coordination complexes (SCCs). These can further assemble and form a variety of interlocked structures such as rotaxanes, molecular knots, catenanes, Borromean rings, interlocked cages, etc. Nonetheless, combinations of different SCCs into catenated forms remain scarcely reported, particularly cross-catenanes derived from distinct metallacages.
Significant progress has been made in coordination-driven self-assembly for creating artificial receptors that mimic biological macromolecules. However, simulating interactions between various biological macromolecules through competitive coordination assemblies remains a nascent field. The primary synthetic hurdle lies in exerting precise control over SCC dynamics during hierarchical assembly, compounded by the challenge of isolating and characterizing low-symmetry end products.
Herein, we successfully synthesized a unique cross-catenane using two positionally isomeric metallacages. The solid-state structure of the cross-catenane was characterized by single-crystal X-ray diffraction, while the dynamic library of self-catenated [2]catenanes and cross-catenanes in solution was studied using variable-temperature NMR experiments and time-of-flight mass spectrometry. To gain a deeper understanding of the energetics involved in the catenated assembly, we employed the TGMin (v.3) program to search for the global-minimum (GM) structures of three [2]catenanes. Density functional theory (DFT) calculations with the B3LYP hybrid exchange-correlation functional were used to calculate the binding energies, revealing that the cross-catenane (-77.09 kcal/mol) is significantly more thermodynamically stable than the self-catenated [2]catenanes (-36.79 kcal/mol and -39.49 kcal/mol). Thus, precise control over the crystallization of low-symmetric cross-catenated metallacages is achieved through structural modulations of SCCs in hierarchical self-assembly.
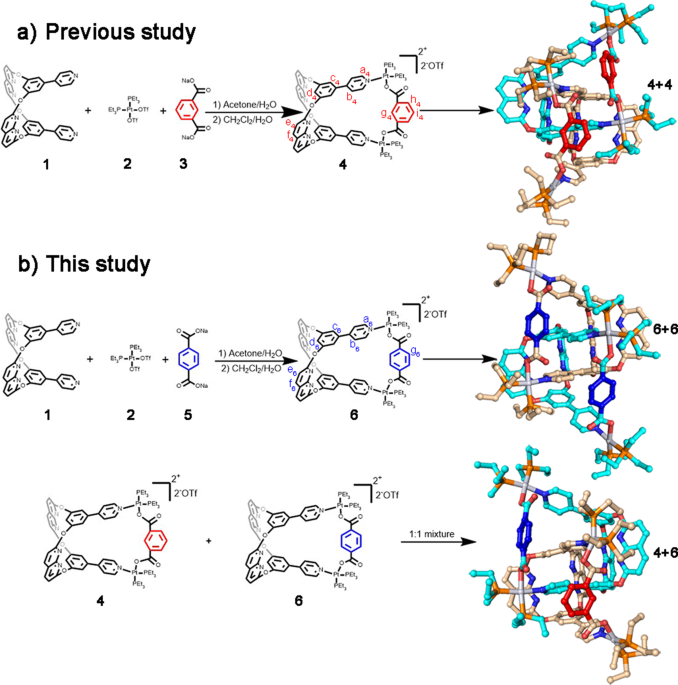
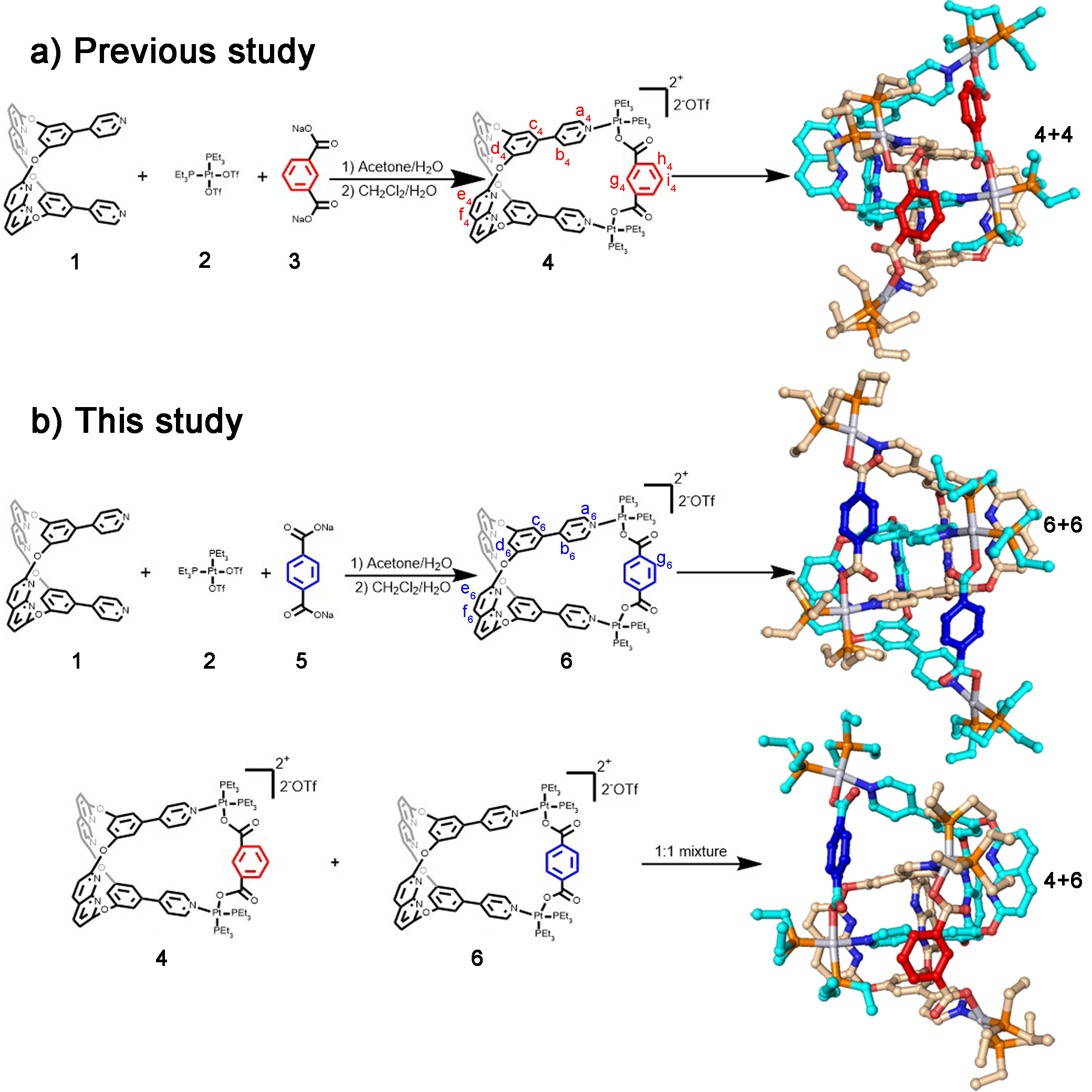 Figure 1 | Synthesis and single-crystal structures of metallacages and [2]catenanes.
Figure 1 | Synthesis and single-crystal structures of metallacages and [2]catenanes.
In this study, we choose two platinum(II) metallacages 4 & 6, containing internal supramolecular binding sites that form [2]catenane respectively in acetone (Figure 1a & 1b). Notably, 6 is a positional isomer of 4, and they share the same internal supramolecular interaction binding sites derived from ligand 1. We expected a slight difference in cavity size between the two metallacages because of the positional isomeric dicarboxylate ligands. In this study, structural differences between the two metallacages were minimized by synthetic design. We use these two metallacages to simulate biological macromolecules, and to use cross-catenation behaviors between metallacages to simulate the interactions between biological macromolecules.
The catenation within the mixed system containing these two metallacages poses a significant challenge to studying them due to their similarity. The two positional isomeric metallacages were used to elucidate catenation behaviour beyond the skeletons’ match, resulting in a cross-catenane in the solid state (4+6, Figure 1b). We dissolved both 4 and 6 at a mole ratio of 1:1 in acetone, and isopropyl ether was slowly added to obtain single crystals. In the crystal structure, 4 and 6 co-existed in the asymmetric unit. After performing a symmetry operation, we observed only cross-catenane 4+6 (Figure 1b). We observed multiple intermolecular interactions between 4 and 6 in cross-catenane 4+6, suggesting that the main driving forces of catenation are the C-H…N bonds between benzene-pyridine arms and naphthyridines, and π···π interactions between naphthyridines.
To verified the generation of cross-catenane in solution, we used 1H NMR experiments to characterise cross-linking in solution. When the concentration of 4 and 6 in the 1:1 mixture in acetone-d6 solution was gradually increased, several broad peaks were observed from 5.0 to 9.0 ppm in the 1H NMR spectra. We expected this based on the complex chemical environment that exists in 4+6. Then, we investigate the stimuli-response of this hierarchal assembled system, we conducted 1H & 31P{1H} NMR (5 mM for each) spectra in acetone-d6 at different temperatures. As shown in Figure 2a, the 31P{1H} NMR & 1H NMR spectra clearly shown signals belongs to the cross-catenane 4+6 at high temperature (pink peaks), while the chemical shifts of 4+4 & 6+6 are occurred due to the temperature variation. However, as the temperature decreased, the signals of cross-catenane were disappeared, and the spectra indicated two self-catenanes (4+4 and 6+6) are the main product in the mixture. This suggests that the dynamic library of catenanes is influenced by temperature, indicating that the cross-catenane 4+6 is more thermodynamically stable than self-catenanes. That is, heating disrupts the self-catenanes, resulting in the formation of more thermodynamically stable cross-catenanes.
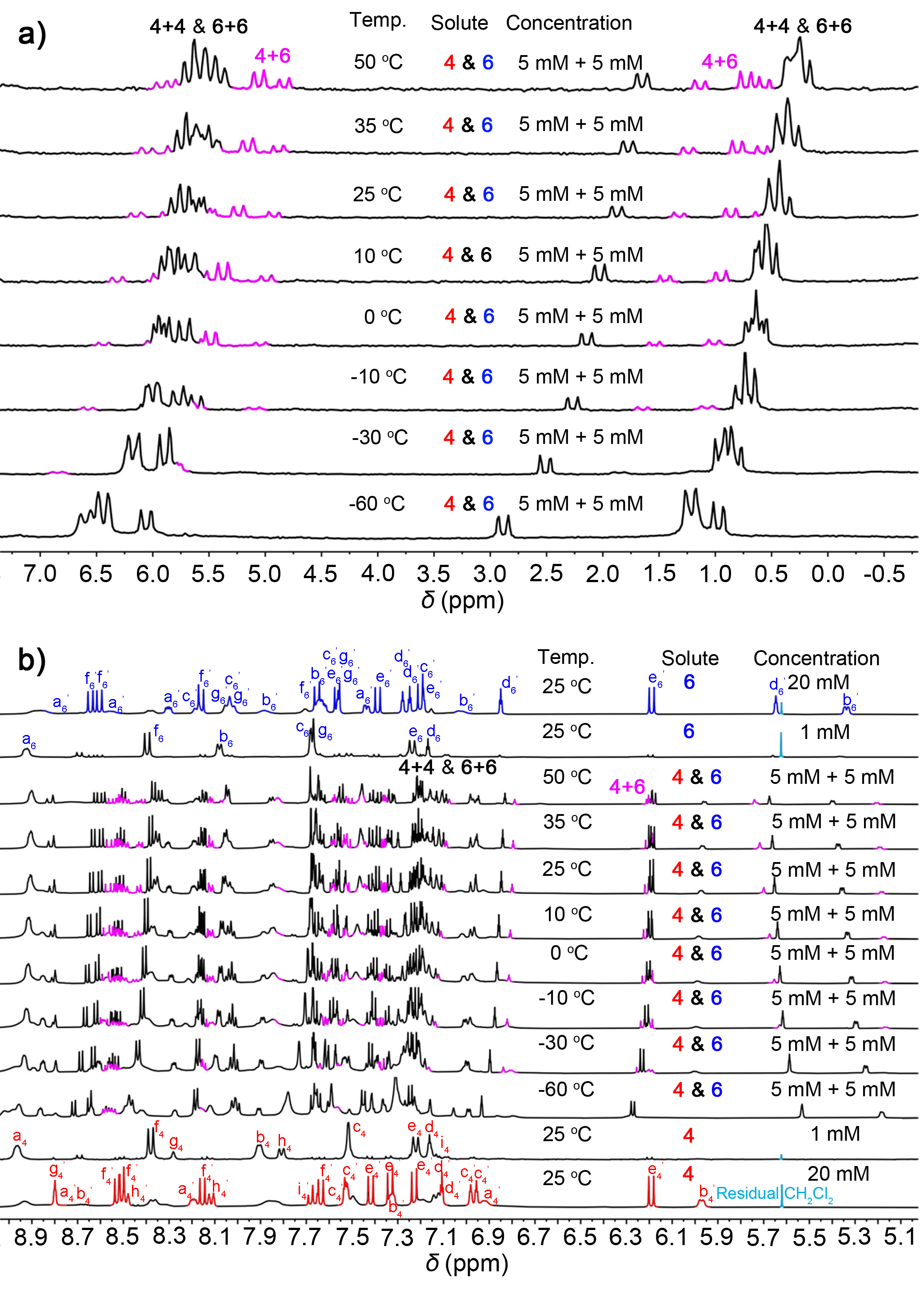 Figure 2 | Varied temperature NMR experiments.
Figure 2 | Varied temperature NMR experiments.
To further describe cross-catenation in solution, metallacage 8 was synthesised based on metallacage 4. As shown in Figure 3a, we introduced a methyl group to the metallacage framework through a dicarboxylated ligand, which could provide high-field proton NMR signals for metallacages and catenanes, and extra molecular weight to identify cross-catenanes in mass spectra. we investigated cross-catenation between 6 and 8 in an acetone-d6 solution. As shown in Figure 3b (entry 3), when the concentration of the 1:1 mixture of 6 and 8 in acetone-d6 solution was gradually increased, an extra proton signal at δ = 2.60 ppm was clearly visible. Thus, we observed the formation of cross-catenane 6+8 in solution, with a conversion of 16.61% at a total concentration of 20 mM (Supplementary Table 7). Moreover, ESI-TOF-MS data also indicated the formation of cross-catenane 6+8 in the mixture, and the results were in good agreement with theory ([M – 2OTf-]2+, Figure 3d).
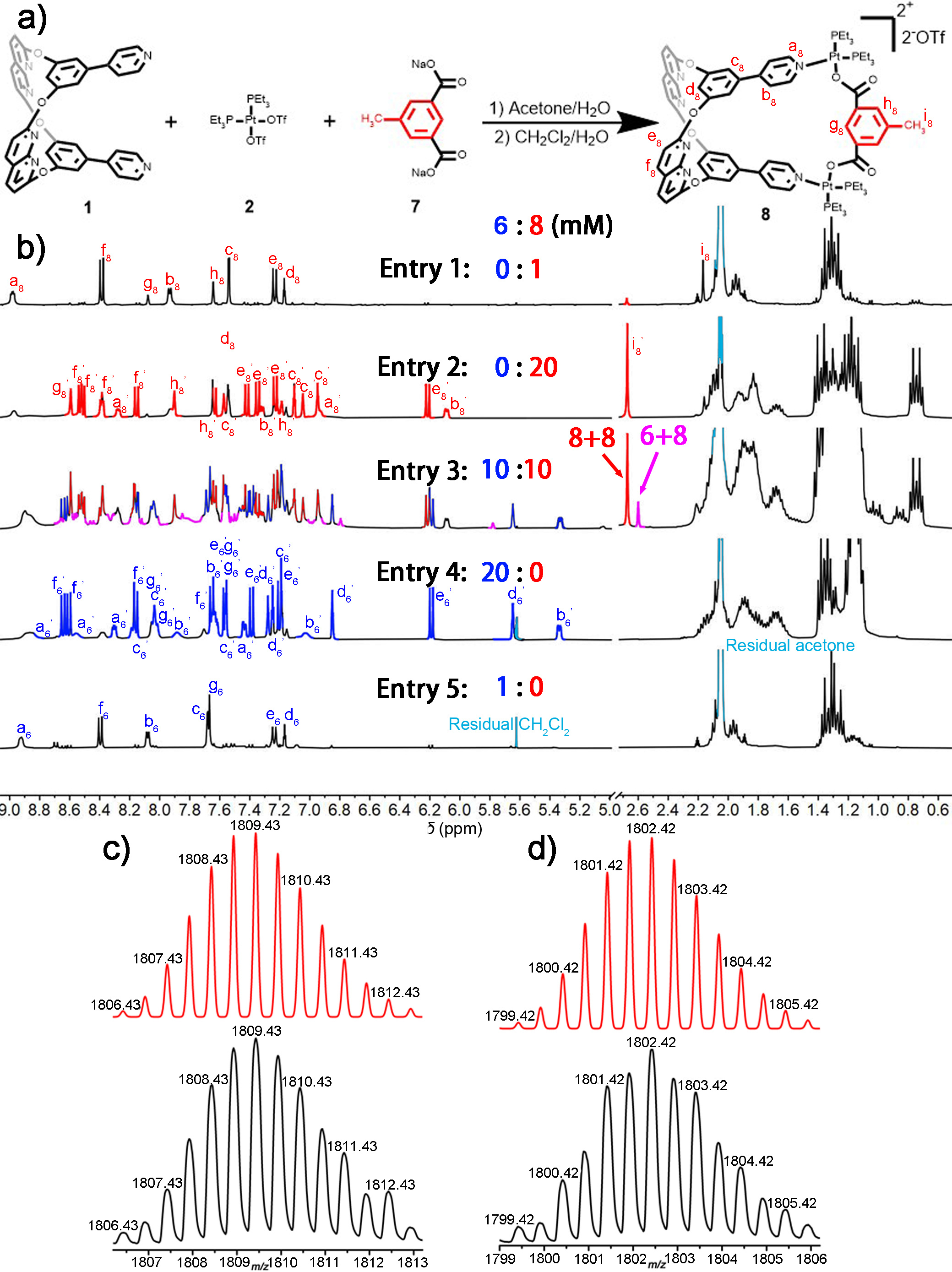 Figure 3 | Synthesis and structural characterize of metallacage 8 and cross-catenane 6+8.
Figure 3 | Synthesis and structural characterize of metallacage 8 and cross-catenane 6+8.
To evaluate the stabilities and mechanisms of cross-catenation, TGMin (v.3) program was employed to search the GM structures of three catananes. In total, 67320 structural isomers of 4+4, 54589 isomers of 4+6, and 55020 isomers of 6+6 were searched, respectively, among all the possible structures. Subsequently, the GM structures of 4+4, 4+6 and 6+6 were re-optimized by using B3LYP exchange-correlation functional in Gaussian program (Figure 4).
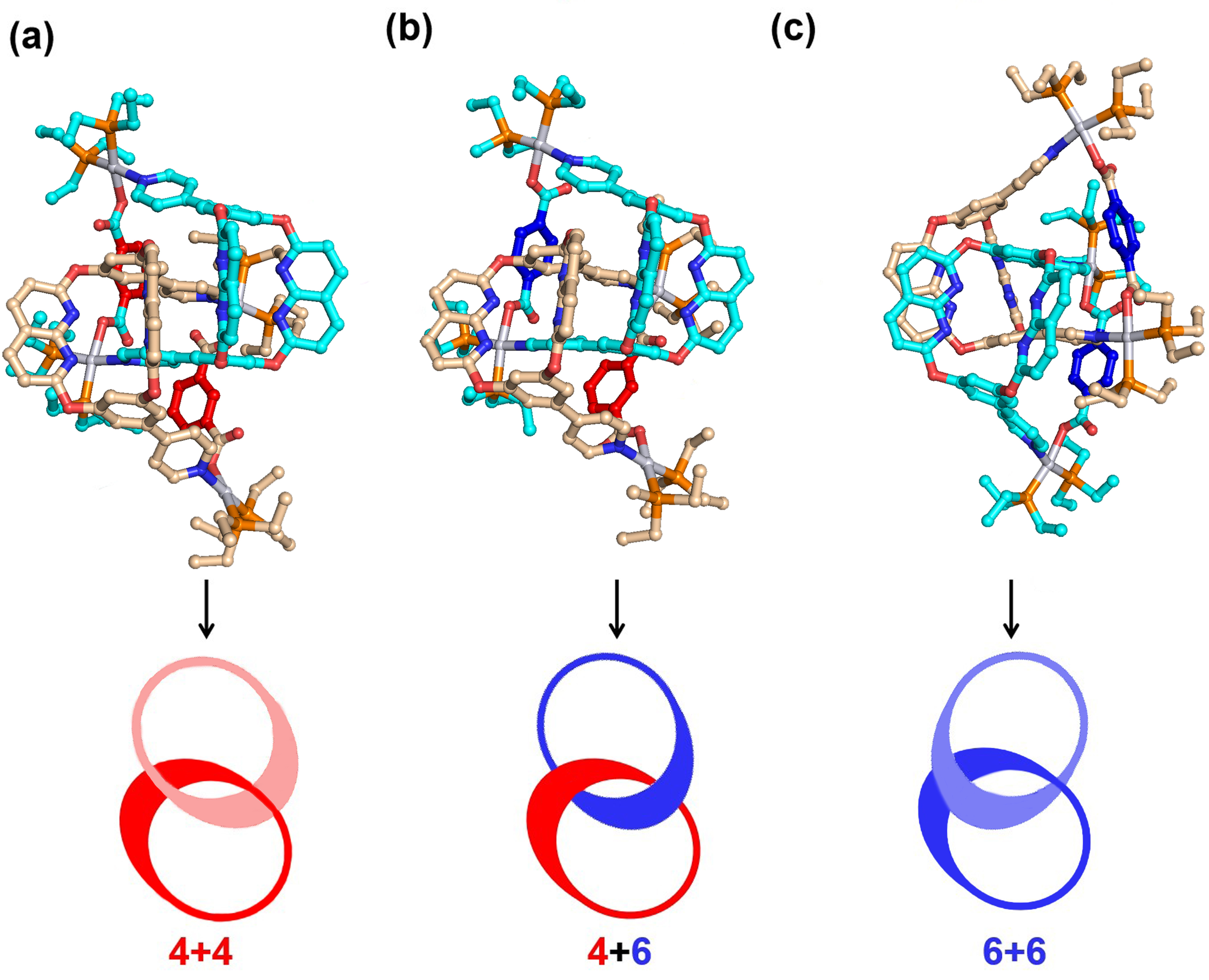 Figure 4 | The global-minimum geometrical structures of [2]catenanes.
Figure 4 | The global-minimum geometrical structures of [2]catenanes.
To account for the experimental results that only 4+6 exists in the mixed single crystals, we have calculated the binding energy (BDE) of [2]catenanes 4+4, 4+6 and 6+6 from two respective monomers. As demonstrated in Table 1, the stabilization energies due to dimerization lie in the order of 4+6 » 6+6 > 4+4. The significantly large BDE of 4+6 indicates the mixed single crystals prefer formation of this 4+6 structure instead of the 4+4 and 6+6 structures. Meanwhile, the mathematical probability of the cross-catenation state (2/3) in a mixed system containing two metallacage 4 and two metallacage 6 is higher than that for the self-catenation state (1/3), indicating that the formation of cross-catenane 4+6 is entropically favorable in crystallization. This is because the minimal structural differences within the metallacages' frameworks strongly influence the hierarchical catenated assembly. Consequently, unique cross-catenated metallacages are preferentially formed in the crystal.
Table 1. The calculated binding energy (BDE, in kcal/mol) of [2]catenanes 4+4, 4+6 and 6+6 from two monomers calculated at the DFT B3LYP/def2-TZVP level of theory.
|
Compound |
Reaction |
BDE |
DBDE |
|
4+4 |
4 + 4 → 4+4 |
-36.79 |
40.3 |
|
4+6 |
4 + 6 → 4+6 |
-77.09 |
0 |
|
6+6 |
6 + 6 → 6+6 |
-39.49 |
37.6 |
In conclusion, we reported the unique example of a cross-catenane obtained from positional isomeric metallacages with single-crystal structures. The solid-state structure of the cross-catenane was characterized by single-crystal XRD, and the dynamic library of catenanes in solution was characterized by NMR and TOF-MS spectra. We conducted global-minimum structure searching and DFT calculations to elucidate the formation mechanism, revealing that the binding energy of the cross-catenane is significantly larger than that of self-catenanes, and thermodynamic driving forces mainly control the selectivity in crystallization. Our findings demonstrate that minimizing structural modulation of SCCs can precisely control their hierarchical interlocked behaviors during crystallization. This result offers facile insight into the future development of low-symmetry/high-complexity supramolecular interlock systems. Further investigations of additional examples are currently underway.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in