Thanks to the revolution of high-throughput sequencing, there are now hundreds of thousands of genomes stored in public databases. Homologous amino-acid sequences contain rich information about how proteins have evolved over millions of years. Can we leverage this data about long-term evolution to predict which amino acids will mutate in the short term?
That’s the key question we had in mind when approaching the “Deciphering polymorphism in 61,157 Escherichia coli genomes via epistatic sequence landscapes“ project.
Understanding protein evolution
Organisms that have evolved independently for millions of years still share homologous proteins. Through long-term evolution, they have fixed so many mutations that, when comparing homologous protein sequences found in two distant species, we often observe sequence identities as low as 20 or 30%. However, these proteins continue to perform the same function and fold the same way in 3D. While evolution has permitted the compilation of many mutations, it has also preserved the key features of the protein.
Let’s consider a simple example (Figure 1). If a residue, such as the one in yellow, is conserved throughout the alignment of homologous protein sequences, it is likely to be crucial to ensure the functionality of the protein. Over millions of years of evolution, whenever a mutation has occurred there, it has been eliminated by natural selection. Therefore, it is unlikely to ever be a polymorphic site, as any amino-acid change would have a detrimental effect on the functionality of the protein.
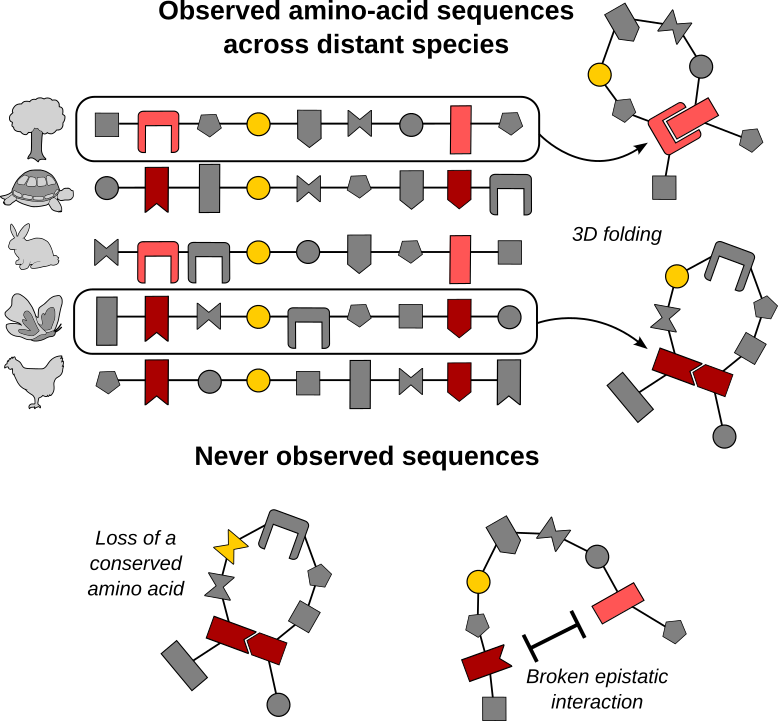
However, a protein is a complex system where residues do not evolve independently from each other. On the contrary, physical and genetic interactions among amino acids - called epistatic interactions - shape the structure, function, and evolution of proteins (Figure 1, red residues). Epistasis makes predicting evolution a daunting task. Indeed, the same amino acid can be beneficial in one sequence - like a dark red residue in a sequence where the partnering residue is also a dark red one - but deleterious in another, for instance if it is facing a light red residue. In other words, the effect of a mutation depends on the genetic context.
But evolution leaves a statistical signature in sequence data. Functionally or structurally important residues are highly conserved (conservation patterns) and, due to epistatic interactions, a subset of amino acids may co-appear with a different frequency than what would be expected based on conservation of the respective residues (covariation patterns).
Our idea is to train a data-driven model that exploits the statistical patterns in the sequence data of distant homologs to predict which amino-acid sites are more likely to be polymorphic in the short term.
Epistasis shapes evolutionary trajectories
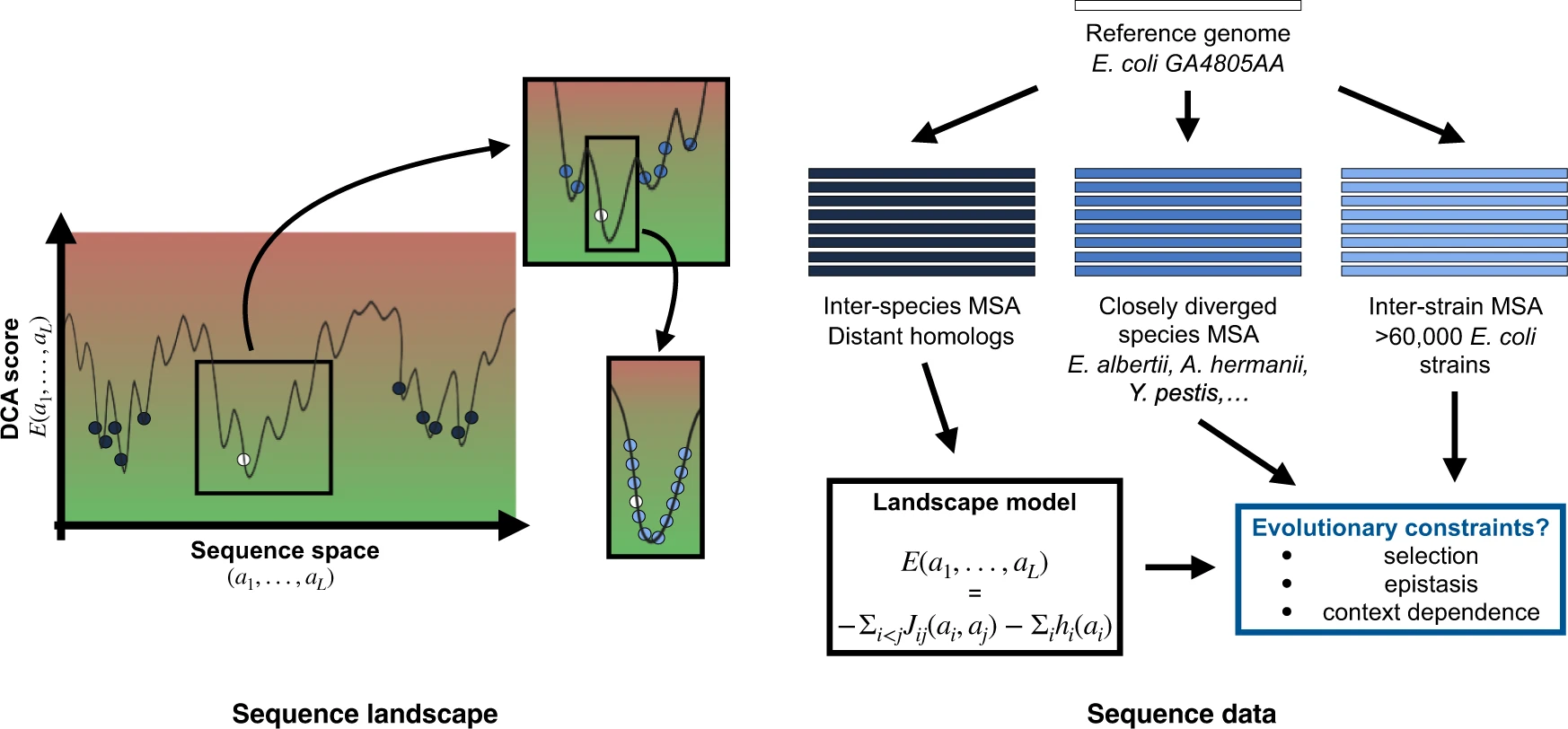
Our genome-scale approach is based on Direct-Coupling Analysis (DCA) [for a short explanation of DCA and a summary of its most successful applications see the https://en.wikipedia.org/wiki/Direct_coupling_analysis]. DCA is an epistasis-aware method that aims at modeling the conservation and covariation patterns in sequence data by explicitly parameterizing them in terms of epistatic couplings and conservation biases.
By comparing our predictions with those of a non-epistatic model (we call it independent model or IND), we prove that accounting for epistasis is crucial to accurately identify the sites where polymorphisms segregate.
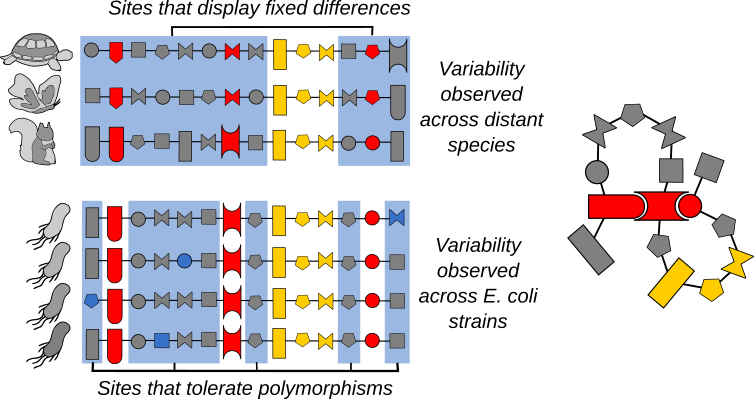
While both the IND and DCA model can predict highly polymorphic sites in the 61,157 Escherichia coli strain, only the epistatic-aware DCA model is able to identify sites that are constrained (i.e. unlikely to mutate) in a specific genetic context. This concerns a large part of the genome: according to our analysis, in 30% to 50% of the sites, the genetic context strongly limits tolerable amino acids. These are sites that can fix differences on long evolutionary times by coevolving with their epistatically interacting partner sites. Therefore, they cannot mutate independently of the rest of the sequence, so they are rarely polymorphic in the short term (Figure 2).
The genetic context: a dense network of epistatic interactions
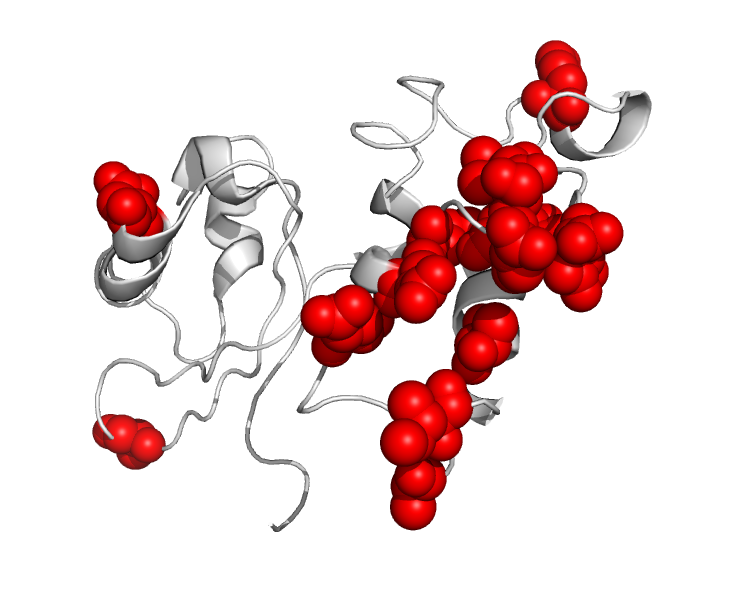
Our work shows that DCA accurately captures most of E. coli’s genetic context. By studying the sequences of species that are closely related to E. coli, we also suggest that genetic context does not rely on rare strong interactions between a few residues but on a dense network of many small couplings. In particular, residues that are not in direct physical contact with one another but contribute to the stability of the same domain in the 3D fold of the protein seem to be epistatically coupled. For instance, when comparing the amino-acid sequence of the L11 protein of 50S subunit of the ribosome in E. coli and in Yersinia pestis, we observe a strong epistatic signal: the amino acids fixed in Y. pestis are far more adapted to Y. pestis’s background than to E. coli’s one. When looking at the 3D structure of the protein, we observe that most of the fixed differences cluster together (red spheres, Figure 3).
In general, clear epistatic signals start to emerge at approximately 10% divergence in amino-acid sequence. This means that in a species like E. coli where the average pairwise diversity is far smaller, the effect of the same amino-acid change in the genetic backgrounds of two different strains should be very similar.
Take-home message
With our work, we have shown that statistical patterns in sequence data of distant homologs (conservation and epistatic patterns) can be exploited to predict polymorphic sites in a set of recently diverged strains.
We proved that this approach was informative of E. coli local genetic context and short-term evolution. This genetic context is crucial to predict the effect of mutations, and epistatic interactions strongly constrain 30%-50% of all amino-acid sites and limit their ability to tolerate polymorphisms. Moreover, our work suggests that the genetic context changes gradually with sequence divergence by accumulating many small couplings.
We took advantage of the large availability of E. coli strain sequence data to test our predictions and validate our approach, but in principle it can be extended to any species for which enough sequence data are available. Indeed, in another project we use a closely related approach to predict which SARS-CoV-2 sites are more likely to mutate in future strains.
In one sentence: looking at the past - protein sequences of distant homologs resulting from millions of years of evolution in different species - can help us predict which sites are going to mutate in the future.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in