Deconvolution of clinical variance in CAR-T pharmacology and response
Published in Bioengineering & Biotechnology


Progress over the last decade in the battle against cancer has taught us that a robust, T cell-mediated anti-tumor response is a requisite for curative therapies. The advent of chimeric antigen receptor T cells (CAR-Ts) has proven that such anti-tumor responses can be provided exogenously, using ex vivo manipulated and genetically engineered T cells. Initial success in treating hematological cancers has propelled the field. Scientists are testing novel CAR-T designs in different cancer types and immune disorders, using increasingly complex multi-gene edits, evaluating other immune cell populations as therapeutics, and exploring the use of allogenic cell sources for starting material (such as induced pluripotent stem cells). Ultimately, the field’s holy grail is off-the-shelf cell therapy products, designed and optimized to treat a range of diseases.
However, T cells make unwieldy therapeutics. These ‘living drugs’ proliferate, differentiate, actively traffic between tissues, and interact with host immune systems in complex and poorly understood ways. As a result, the pharmacology of T cells is highly variable. Exposure (circulating CAR-T cell AUC) can vary by three orders of magnitude between patients administered the same ‘dose’, a level unheard of for small molecules or biologics. The biological processes underlying this variance remain poorly characterized, and empirical modelling approaches typically used to quantify pharmacokinetic-pharmacodynamic (PKPD) relationships fall short of providing meaningful insight into this biology.
We hypothesized that the rules governing CAR-T pharmacology could be extrapolated from the dynamics of T cell responses to infection. After extensive literature reviews, discussions with immunologists and many iterations, we devised a relatively simple but elegant mathematical description of T cell regulatory control based on an analogy to a ‘toggle switch’. Antigens expressed on tumor cells function as the switch, coordinating T cell proliferation and differentiation between three canonical T cell states (memory, effector, and exhausted). We trained the model using CAR-T and B cell (tumor) count data from a trial of Kymriah (a CD19-targeted CAR-T) in chronic lymphocytic leukemia, and queried what kinetic parameters underly differences in pharmacology and clinical outcomes. Minimal in vivo expansion and responses were predicted to be caused by cell-intrinsic deficits in the infused CAR-T cell products. Specifically, deficits in the proliferative capacity of memory cells and cytotoxic potency of effectors. We next simulated a virtual clinical trial where variability was encoded in CAR-T product archetype, dose, and tumor burden. Simulations accurately predicted the clinical variance in exposure and covariates of response in additional clinical data. This suggests that additional pharmacokinetic variability results from interactions between the CAR-T dose and patient tumors.
We then sought to translate these predicted cell-intrinsic differences in memory cell proliferation kinetics to measurable biomarkers. We searched for transcriptional signatures of T cell function in bulk and single-cell transcriptome data of CD19 CAR-T products matched with clinical response in different hematological malignancies. We found that memory cell sub-populations from poor responders show heightened expression of a CART dysfunction gene signature, as well as other transcriptional features indicative of diminished function. We identified 28 pathways that significantly differ between CAR-T products associated with robust vs. poor response, and developed a machine learning workflow to predict clinical outcomes based on these pathways. We found that these transcriptional signatures accurately predict clinical outcomes in multiple datasets, and do so better than models trained on immunophenotypes routinely measured and reported to correlate with response.
So why is this important? As we work to create the next generation of T cell therapies with enhanced efficacy, safety, and batch-to-batch consistency, robust predictive models are required to guide product design and treatment (Figure 1). Routine transcriptome profiling combined with our machine learning workflow can be used to evaluate the proliferative capacity of novel cell products. Measurements of remnant tumor burden (B cell counts) from patients following lymphodepleting chemotherapy could then be used as inputs to our dynamical model, and simulations used to design dosing regimens which maximize anti-tumor response while minimizing excess toxicity.
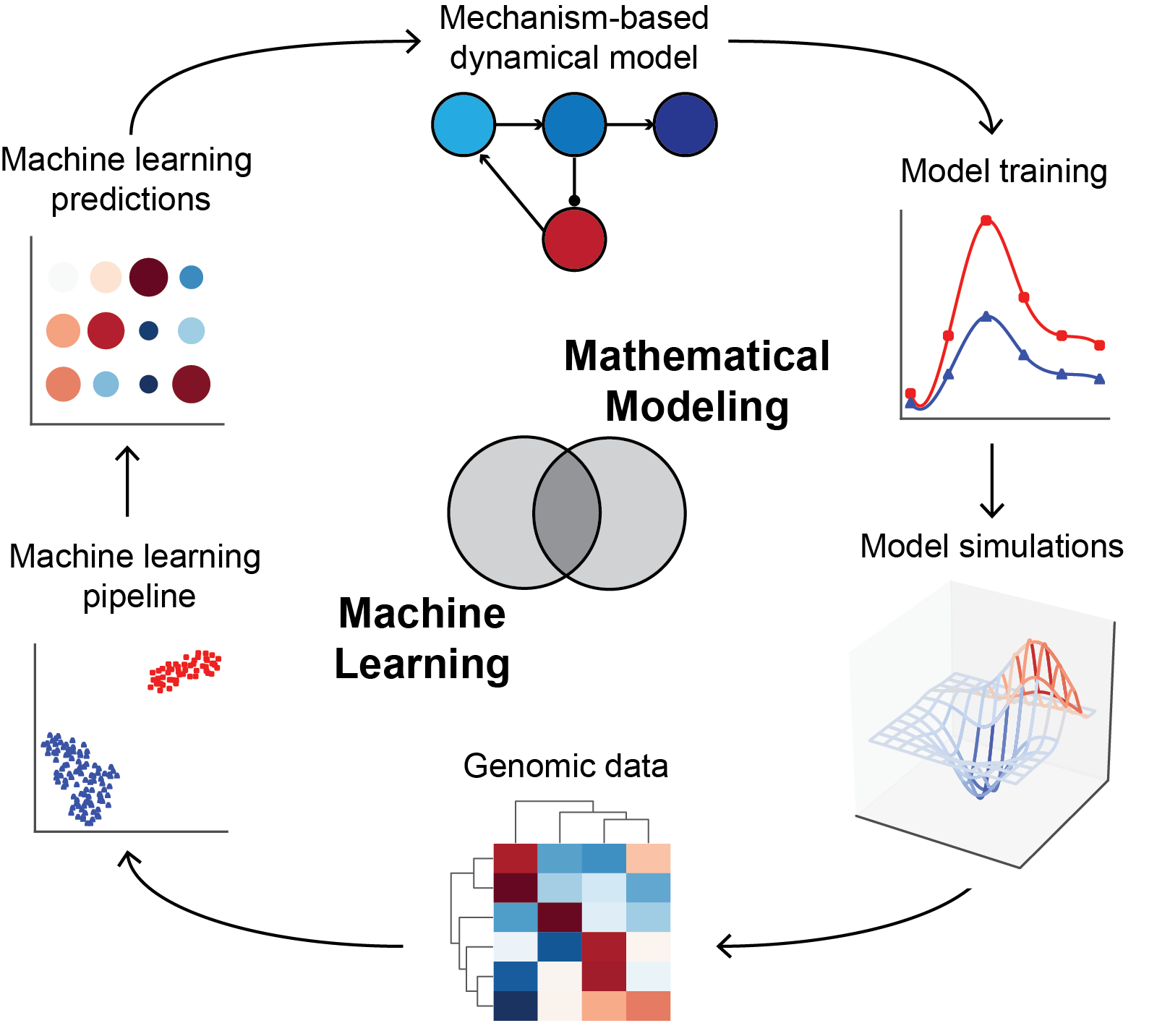
Figure 1: Our approach to integrating mechanism-based mathematical modeling and machine learning. Biological mechanism (both established and hypothesized) informs the development of differential equation-based mathematical models. These are trained on dynamic data such as clinical pharmacokinetics and pharmacodynamics. Insights gained from model simulations can be assessed using genomic data, and machine-learning approaches then used to extend insights into novel biology. Machine learning predictions are translated into testable predictions, which inform the next generation of models and experiments in an iterative cycle.
Building on this work will require additional clinical data for model training, testing and refinement. Hundreds of novel CAR-T clinical trials are in progress – opening the underlying data up to the larger scientific community could usher in a new wave of insights and understanding, fueled by bright computational scientists around the globe. To develop the ‘holy grail’ of accessible, safe, and effective cell therapies, we will need all the help we can get.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in