Direct time-resolved observation of surface-bound carbon dioxide radical anions on metallic nanocatalysts
Published in Chemistry
The chemical and physio-chemical conversion of CO2 is a crucial global issue. However, the efficacy is still limited by an incomplete understanding of elementary steps, especially the reaction between CO2•‒ radicals and nanocatalysts as one of the reaction-determining steps1,2. Direct time-resolved identification of the species on metallic nanocatalysts is imperative but challenging because the existing in-situ spectroscopic techniques such as Infrared (IR), Raman, and X-ray Absorption Spectroscopy only provide subsecond time-resolution.
To fill this gap, we develop a direct, time-resolved observation of the elementary reaction between CO2•‒ radicals and metallic nanocatalysts in aqueous solution. Water radiolysis produces quantitative hydrated electron (eaq–), known as “electrolysis without electrode”. The picosecond pulse radiolysis coupled with transient absorption spectroscopy (ELYSE platform, Paris-Saclay University) could form CO2•‒ radical anions via eaq– attachment to aqueous CO2 with an almost diffusion-controlled rate (8 × 109 M-1 s-1) within 10 nanoseconds3,4. The characterization of transient absorption profiles can further disclose the electronic transition of surface-bound intermediates.
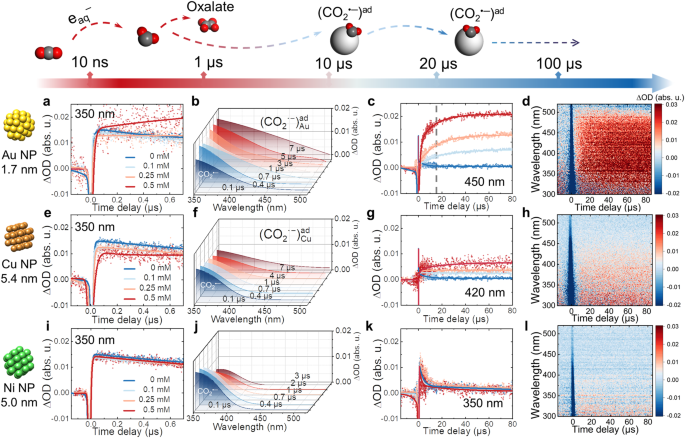
, 3D stereograph of fitted transient absorption spectra within 7 µs in 0.5 mM Au solution (b), transient kinetics at 420 nm as a function of Au concentration (c), and transient absorption matrix (d) within 80 µs in 0.5 mM Au solution. e-h, Transient kinetics at 350 nm within 700 ns as a function of Cu concentration (e), 3D stereograph of fitted transient absorption spectra within 7 µs in 0.5 mM Cu solution (f), transient kinetics at 450 nm as a function of Cu concentration (g), and transient absorption matrix (h) within 80 µs in 0.5 mM Cu solution. i-l, Transient kinetics at 350 nm within 700 ns as a function of Ni concentration (i), 3D stereograph of fitted transient absorption spectra within 7 µs in 0.5 mM Ni solution (j), transient kinetics at 350 nm as a function of Ni concentration (k), and transient absorption matrix (l) within 80 µs in 0.5 mM Ni solution.")
Fig. 1 │ Time-resolved absorption of CO2•‒ radical stabilization process with different metal NP. a-d, Transient kinetics at 350 nm within 700 ns as a function of Au concentration (a), 3D stereograph of fitted transient absorption spectra within 7 µs in 0.5 mM Au solution (b), transient kinetics at 420 nm as a function of Au concentration (c), and transient absorption matrix (d) within 80 µs in 0.5 mM Au solution. e-h, Transient kinetics at 350 nm within 700 ns as a function of Cu concentration (e), 3D stereograph of fitted transient absorption spectra within 7 µs in 0.5 mM Cu solution (f), transient kinetics at 450 nm as a function of Cu concentration (g), and transient absorption matrix (h) within 80 µs in 0.5 mM Cu solution. i-l, Transient kinetics at 350 nm within 700 ns as a function of Ni concentration (i), 3D stereograph of fitted transient absorption spectra within 7 µs in 0.5 mM Ni solution (j), transient kinetics at 350 nm as a function of Ni concentration (k), and transient absorption matrix (l) within 80 µs in 0.5 mM Ni solution.
We combined the time-resolved method with molecular simulations to identify the surface-bound intermediates for three typical metallic nanocatalysts: Cu, Au, and Ni. The characteristic transient absorption spectra and distinct kinetics from nanosecond to the second timescale reveal how the nature of the metallic nanocatalysts influences the stabilization of the radical on its surface. The surface of Au and Cu has substantially extended the lifetime of [CO2•−]ad radicals by at least 100 times compared to CO2•‒ radicals decayed in bulk solutions or in the presence of Ni. The extended lifetime makes the subsequent occurrence of multi-electron transfer reactions (Figure 1). At a sub-second range, we observed the dimerization pathway of surface-bound CO2•− radicals solely on Cu, which explains the unique capability of Cu to produce C2H4 (Figure 2). These observations revisit the principle of CO2 selective catalysis from a transient perspective.

Fig. 2 │Transient absorption profiles at second timescale. a-b, Transient absorption spectra of surface-bound radicals at 1 ms, 50 ms, 150 ms, 500 ms, and 900 ms in the presence of 0.5 mM 5.4 nm Cu (a) and 5.3 nm Au (b). c-d, The curves of ΔO.D. versus time delay at 350 nm (c) and 540 nm (d). e, Schematic diagram of the generation, stabilization, and conversion of CO2•− radicals in the presence of Au and Cu NP. CO2 reacted with eaq– to form CO2•– within a few nanoseconds, and the CO2•− radicals stabilized on the surface of Cu and Au NPs. After 1 ms, surface-bound radicals were converted to dimer intermediates, and surface-bound radicals decayed for around 150 ms. For simplicity, cations are not shown.
Factors such as metallic size and electrolytes are often important to catalytic CO2 reduction. Our experimental results also showed that, in the range of 1.7 ~ 6.6 nm, the smaller Au NP enhances the surface-bound interaction. This is due to more active sites and localized electron distribution among stabilized CO2•− radicals and Au surfaces. Moreover, we found that alkali metal cations facilitate the stabilization of the surface-bound CO2•−, which is in good agreement with electrocatalytic studies. Meanwhile, we proposed that excessive cations would form ion pairs by electrostatic binding to CO2•−. The binding interactions hinder the initial diffusion and interfacial stabilization process. For Li+, Na+, and K+ ions, the larger the cation radius, the more obvious the negative effect. The findings provide direct spectroscopic evidence on catalyst size and alkali metal cations in the electrolyte effects on the first elementary step of CO2 reduction.

Fig. 3 │ The effect of catalyst size and cation in solutions on CO2•‒ radical stabilization process with Au. a-c, Transient absorption spectra of different sizes of Au (0.5 mM) at 75 µs (a) and transient kinetics at 350 nm (b) and 450 nm (c) within 80 µs. d-f, Transient absorption spectra of different sizes of Au (0.5 mM) at 750 µs (d) and transient kinetics at 350 nm (e) and 450 nm (f) within 800 µs. g-i, Transient kinetics at 350 nm with different concentrations of Li+ (g), Na+ (h), K+ (i). j-l, Transient absorption spectra at 750 µs with varying concentrations of Li+ (j), Na+ (k), K+ (l).
This work highlights fundamental ultrafast spectroscopy to clarify the critical initial step in CO2 catalytic reduction. The selective stabilization observation offers a more straightforward insight into the intrinsic mechanism of CO2 reduction and thus reveals the catalyst design principle.
For more detailed information, see our article “Direct time-resolved observation of surface-bound carbon dioxide radical anions on metallic nanocatalysts” in Nature Communications (https://www.nature.com/articles/s41467-023-42936-6), featured by Christian Kuttner to Editors’ Highlights in “Inorganic and physical chemistry” (https://www.nature.com/collections/wtpqpqpgwd).
- Kong, S. et al. Delocalization state-induced selective bond breaking for efficient methanol electrosynthesis from CO2. Nat. Catal. 6, 6–15 (2022).
- Xu, A. et al. Copper/alkaline earth metal oxide interfaces for electrochemical CO2-to-alcohol conversion by selective hydrogenation. Nat. Catal. 5, 1081–1088 (2022).
- Belloni, J. et al. ELYSE—A picosecond electron accelerator for pulse radiolysis research. Meth. Phys. Res. A 539, 527-539 (2005).
- Marignier, J. L. et al. Time-resolved spectroscopy at the picosecond laser-triggered electron accelerator ELYSE. Phys. Chem. 75, 1024-1033 (2006).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in