Directly elucidate the structure of small molecules from a miniaturized mass spetrometry
Published in Chemistry
The analysis of compound structures in mass spectrometry detection and the generation of complete molecular structures solely from tandem mass spectra have been longstanding goals in analytical chemistry. Achieving accurate structure predictions holds immense potential in various fields such as discovering new homologous derivatives, natural product research, untargeted metabolomics, food safety, drug component analysis, and drug detection. In the field of on-site detection, there is a growing need for rapid, efficient, and accurate detection of molecules, which can be effectively met by on-site mass spectrometers. However, compared to conventional laboratory-scale mass spectrometers, on-site mass spectrometers are constrained by size and performance limitations, resulting in lower spectral data resolution. This significantly restricts their application scenarios, preventing users from extracting relevant information about the measured substances with precision from the spectra.
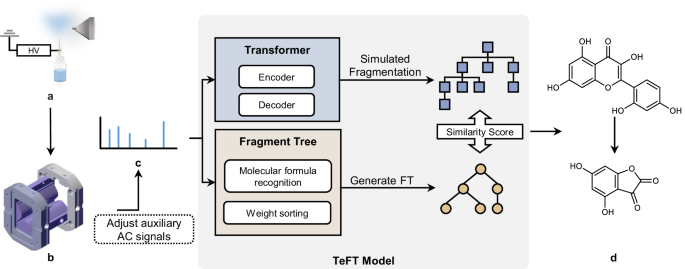
Given the diversity of compound structures and the limitations of on-site mass spectrometers in terms of resolution and accuracy, rapid determination of the structure of the target compound still faces significant challenges. Therefore, it is imperative to establish an identification model based on on-site mass spectrometers and low-resolution spectra to predict the structure corresponding to the spectrum and provide a reference for subsequent analysis. The spectrum recognition model developed in this study includes a deep learning model, Transformer, as well as molecular fragment tree models, namely the fragment tree and SMILES fragment tree models. This hybrid learning model achieves complete prediction of unknown substance structures solely from low-resolution and low-accuracy tandem mass spectra, while also realizing spectrum recognition on on-site mass spectrometers. This model allows for deeper processing of spectra, enabling the recognition of substance structures and their homologous derivatives.
The entire algorithm model consists of the deep learning model, Transformer, the fragment tree model generated through simulated fragmentation (SMILES fragment tree), and the fragment tree model directly generated from the original tandem mass spectrometry data. The workflow of the spectrum recognition algorithm is illustrated in Figure 1. During detection, the target substance is ionized using an electrospray ionization source, and the ions enter the miniature mass spectrometer for detection. By applying auxiliary AC signals to the ion trap via terminal control, isolation and collision-induced dissociation of the target substance are achieved, resulting in the acquisition of its fragmentation spectrum. Subsequently, the algorithm model processes the spectrum data. Initially, the deep learning model, Transformer, predicts a series of potential structures of the target molecules from the tandem mass spectra, represented by SMILES strings. The generated results are then subjected to simulated fragmentation to convert them into a series of SMILES fragment trees. Meanwhile, the original mass spectrum data is transformed into its corresponding molecular fragment tree through a fragment tree generation algorithm, and similarity scores are calculated for each SMILES fragment tree. The SMILES fragment tree with the highest score contains the most likely fragmentation pathways and potential structures during the fragmentation process, achieving structural annotation of the mass spectrum and predicting the structure of the on-site detection substance. This method can be applied to any mass spectrometry system, particularly suitable for on-site mass spectrometers with limited mass resolution and accuracy.
We conducted a series of drug experiments to validate the performance of the spectral recognition model. Twenty-three flavonoid and astragalus compounds were selected for analysis, and the model successfully predicted the complete structures of twelve substances. The experimental results demonstrate that the model exhibits good predictive performance on field mass spectrometers with lower mass resolution and accuracy. It can predict the complete structures of tested substances from tandem mass spectra, with molecular fingerprint similarities exceeding 0.93 in the final predictions. Additionally, the model can provide corresponding structural annotations for some fragmentation peaks in the spectra. Moreover, in the experiment for identifying unknown components in traditional Chinese medicine capsules, the model successfully predicted one of the substances in the Anweiyang Capsules, achieving rapid substance detection and component identification using field mass spectrometers.
Our research enables direct structural prediction of tandem mass spectrometry data with low mass resolution and accuracy, significantly reducing the data processing workflow. We provide algorithmic models for substance detection using field mass spectrometers, thereby broadening the application scenarios of such instruments.

Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in