DNA copy number profiling at single-cell resolution
Published in Bioengineering & Biotechnology
Tumors are complex ecosystems composed of billions of cells that constantly evolve. Intra-tumor heterogeneity (ITH) refers to the diversity of cancer cells within a single tumor mass. This diversity can manifest in various ways, including differences in genetic mutations, gene expression patterns, cellular morphology, and metabolic activity. ITH is associated with disease prognosis and the risk of developing resistance to drug therapies, which presents significant challenges for both doctors and scientists.
DNA copy number alterations (CNAs) are a common feature of cancer genomes. CNAs involve gains or losses of segments of DNA, leading to an imbalance in the copy number of genes within the affected regions. These alterations can encompass whole chromosomes, chromosomal arms, or smaller genomic segments and contribute to the genetic diversity observed within a tumor. As propagating genetic traits, CNAs can drive clonal evolution by providing a selective advantage to cancer cells with specific genomic alterations. Cells harboring CNAs that confer resistance to therapy or promote aggressive phenotypes may expand within the tumor population over time, leading to the emergence of diverse subclones. Therefore, understanding CNAs allows for the depiction of the ITH portraits as well as the inference of tumor evolution trajectories.
CNAs can be detected in clinical samples using either array-based techniques, such as array comparative genome hybridization (aCGH) and single-nucleotide polymorphisms (SNP) arrays, or sequencing-based techniques, such as whole genome sequencing (WGS) or whole exome sequencing (WES). However, traditional bulk sequencing methods provide an average representation of the genomic landscape across all cells in a sample, masking the diversity of CNAs that may exist among individual cells within the tumor or tissue. Developing single-cell CNA profiling technologies represents a crucial step toward unraveling the complexity of tumor heterogeneity and translating this knowledge into more effective strategies for cancer diagnosis, prognosis, and treatment.
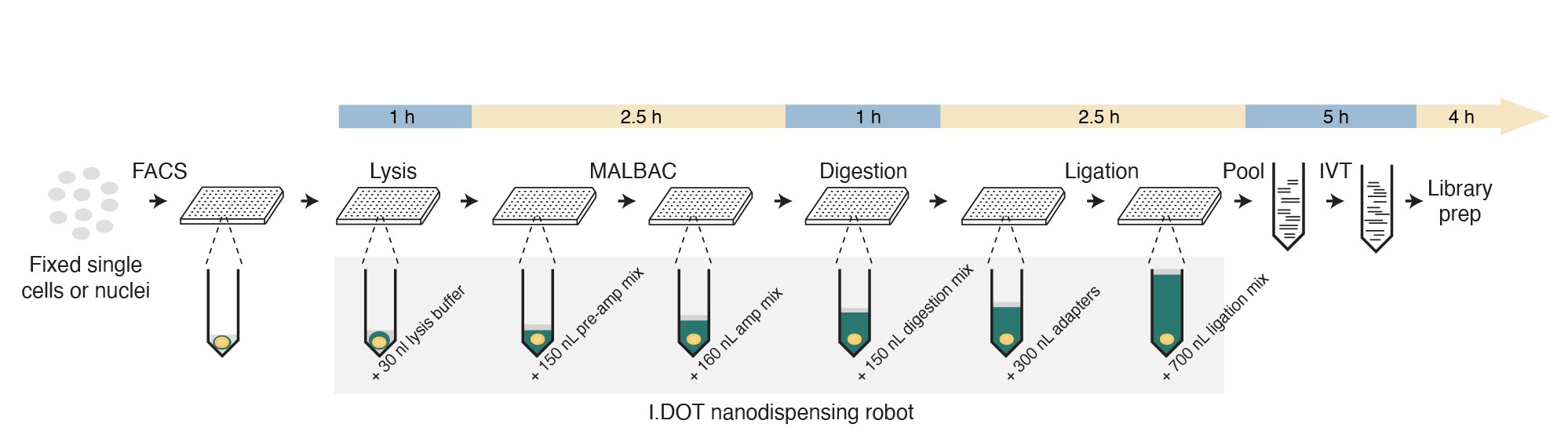
Motivated by the need to profile CNAs in tumors with single-cell resolution, back in 2019 we decided to develop a single-cell version of our previously described CUTseq method (10.1038/s41467-019-12570-2) that would allow us to profile CNAs in multiple single cells in a cost-effective manner. We now describe this new method, which we name scCUTseq, in an article published in Nature Communications (10.1038/s41467-024-47664-z).
In brief, we first sort single cells or nuclei in 384 well plates that are pre-filled with mineral oil and employ Multiple Annealing and Looping-Based Amplification Cycles (MALBAC) to pre-amplify genomic DNA in each well. We then digest the pre-amplified genomic DNA with a single restriction enzyme that cuts DNA at thousands of defined locations. Afterwards, the DNA fragments are ligated with oligonucleotide adapters that contain, among others, a sample-specific barcode sequence and the T7 promoter sequence. After ligation, up to 384 single-cell samples are pooled together, and the barcoded genomic DNA is linearly amplified using in vitro transcription. Finally, a library is prepared from the resulting amplified RNA and sequenced on Illumina platforms. After sequencing, each read is assigned to a unique single cell based on the barcode sequence present at the start of the read. Noteworthy, to minimize the volume of reagents used and reduce the cost per sample, we performed all the reactions before the pooling step in nanoliter volumes using the I.DOT nanodispenser, which we previously deployed for high-throughput CUTseq.
We first assessed whether scCUTseq can detect single-cell CNAs from different cell lines with or without fixing them with formaldehyde. Indeed, our method can resolve the copy number profiles of different cell lines independently of the cell fixation process and is free of cross-contamination, highlighting its specificity. We then assessed the sensitivity of scCUTseq by testing its ability to detect a 7 megabase (Mb) deletion introduced by CRISPR-Cas9 in a small percentage (3%) of cells. It turns out that scCUTseq not only could detect a single copy of the exact 7 Mb deletion in 3.3% cells but also detected 3.2% cells harboring the deletion until the end of the q-arm, which was independently validated by DNA fluorescence in situ hybridization (FISH). We proceeded to benchmark scCUTseq by comparing it to Acoustic Cell Tagmentation (ACT) (10.1038/s41586-021-03357-x), another single-cell CNA profiling method based on DNA tagmentation, which does not require a pre-amplification step. The results revealed that the CNAs detected by scCUTseq closely resembled those identified by ACT and were not artifacts attributable to the MALBAC step.
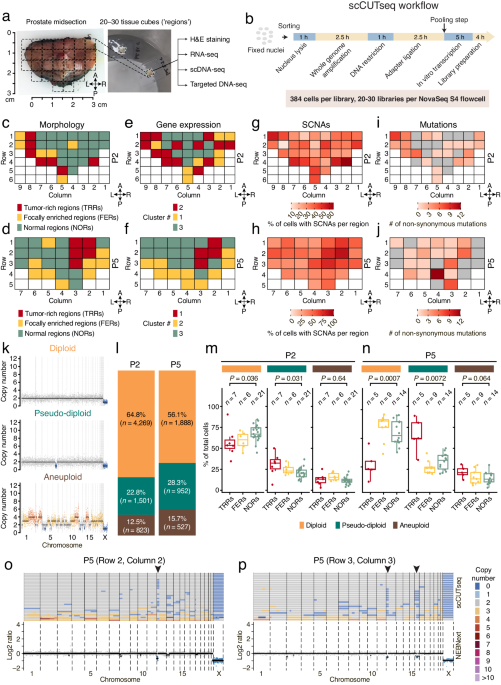
Motivated by these results, we decided to apply scCUTseq to prospectively collected prostatectomy samples from six patients diagnosed with localized prostate cancer. Each transversal prostate midsection sample had already been sectioned into multiple small tissue blocks (~125 cubic mm, hereafter named ‘regions’). Based on histopathological examination, each region was categorized as either tumor-rich regions (TRRs, containing >50% tumor cells), focally enriched regions (FERs, with 10–50% tumor cells), or normal regions (NORs). We then systematically profiled CNAs across all the tissue regions (~30 regions per patient) from two prostate patients (P2 and P5) with scCUTseq, generating DNA copy number profiles from 23,808 nuclei from 62 regions in total. Subsequently, we trained a random forest classifier to filter out low-quality profiles, resulting in the retention of 9,960 cells for further analysis. Our analysis revealed a widespread spatial distribution of cells exhibiting CNAs across all regions from P2 and P5, with a notable enrichment of CNAs within TRRs and FERs. Additionally, parallel targeted deep DNA sequencing for each region showed that point mutations were more localized in TRRs and FERs compared to CNAs, with some also detected in NORs. In contrast, scCUTseq on fresh frozen human forebrain and skeletal muscle autopsy samples revealed a minimal number of CNAs, providing further validation of the reliability of CNAs detected by scCUTseq.
Upon further visual inspection, we revealed three distinct cell populations: diploid cells exhibiting no alteration, pseudo-diploid cells harboring a few sparse alterations, and aneuploid cells characterized by multiple whole-chromosome alterations. Among these cells, diploid cells were significantly abundant in NORs, as expected. Conversely, pseudo-diploid cells were found significantly enriched in TRRs while aneuploid cells were evenly distributed among TRRs, FERs, and NORs. Prompted by these observations, we wondered whether the pseudo-diploid cells detected are clonally related and form spatially distinct subclones. Phylogenetic analysis identified the existence of numerous highly divergent subclones (n = 79 and 52 in P2 and P5, respectively), a surprising finding compared to the substantially fewer subclones identified through scCUTseq on two breast cancer samples (n = 7 and 3, respectively). To further explore the spatial distribution of these subclones, we introduced the Shannon entropy index. Our analysis indicated a subset of highly localized subclones (with low index) harboring mainly pseudo-diploid cells were concentrated in tumor-rich regions. To validate our findings, we employed an orthogonal method, DNA FISH, to visualize the spatial distribution of a pseudo-diploid cell subclone featuring a monoallelic sub-chromosomal arm deletion. This deletion was consistently detected by both scCUTseq and ACT, reaffirming the reliability of scCUTseq as a single-cell DNA copy number profiling method.
In conclusion, we have developed a versatile and cost-effective method for high-throughput single-cell DNA copy number alteration profiling, enabling the depiction of genetic ITH within tumors. Our approach can be used to unveil the phylogenetic evolution of the tumor subclones and to detect the set of high-risk CNAs that contribute to tumor initiation and progression. Notably, scCUTseq is especially suited for profiling CNAs in single cells and nuclei extracted from patient-derived samples. Stabilization of cells or nuclei through fixation allows storing them for several weeks up to a few months before sorting. Furthermore, up to 384 single cells can be multiplexed in a single library, offering flexibility both in time and sequencing. We anticipate that scCUTseq could also be applied to nuclei extracted from formalin-fixed paraffin-embedded (FFPE) tissue samples since it builds on the CUTseq method, which we previously specifically tailored for these samples.
So, if you want to use scCUTseq and need expert advice, please contact the developer of scCUTseq, Ning Zhang (zhangning0816@gmail.com). We look forward to helping you!
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in