DTLL overcomes chemoresistance in PDAC through different SMAD4-mediated mechanisms
Published in Cancer

Pancreatic ductal adenocarcinoma (PDAC) is the most aggressive and fatal cancer, with a five-year survival of approximately 6-9%. Chemoresistance to gemcitabine limits its clinical implementation for pancreatic ductal adenocarcinoma (PDAC) with profound symptomatic effects, although gemcitabine is still the first-line treatment. Additional therapeutic strategies have been explored, especially for drug-resistant PDAC, including resistance to multiple chemotherapies, EGFR-targeted therapies and PD-1/PD-L1 immunotherapies in PDAC. However, the newly developed immunotherapy even showed a poor effect [1-4].
In the current precision medicine treatment era, diverse genetic alterations in cancer with unique hydrogenous signatures are responsible for the therapeutic resistance of pancreatic cancers as one of the biggest challenges. The most common mutations in PDAC occur in KRAS, TP53, CDKN2A, and SMAD4 driver genes. In contrast to other oncogenes (such as mutated BRAF or EGFR), few effective therapies that directly target these four driver genes are available for PDAC. Recent studies have demonstrated that these tumor drivers play roles in gemcitabine susceptibility to pancreatic adenocarcinoma cells [5-10]. However, the approval of sotorasib (Lumakras, a KRAS-G12C inhibitor) by the American Food and Drug Administration (FDA) in 2021 for the treatment of advanced non-small cell lung cancer (NSCLC) encourages more attention to those key drivers that served as undruggable targets previously. In addition, to overcome intrinsic and acquired resistances, researchers are now working on combination strategies. Several combination strategies of either targeted agents or checkpoint inhibitors and chemotherapies showed synergistic activities, highlighting the potential of these strategies for pancreatic cancer [11, 12]. Therefore, a personalized approach targeting key drivers in PDAC with combinational therapy is likely to be a future trend.
What’s novel about our paper
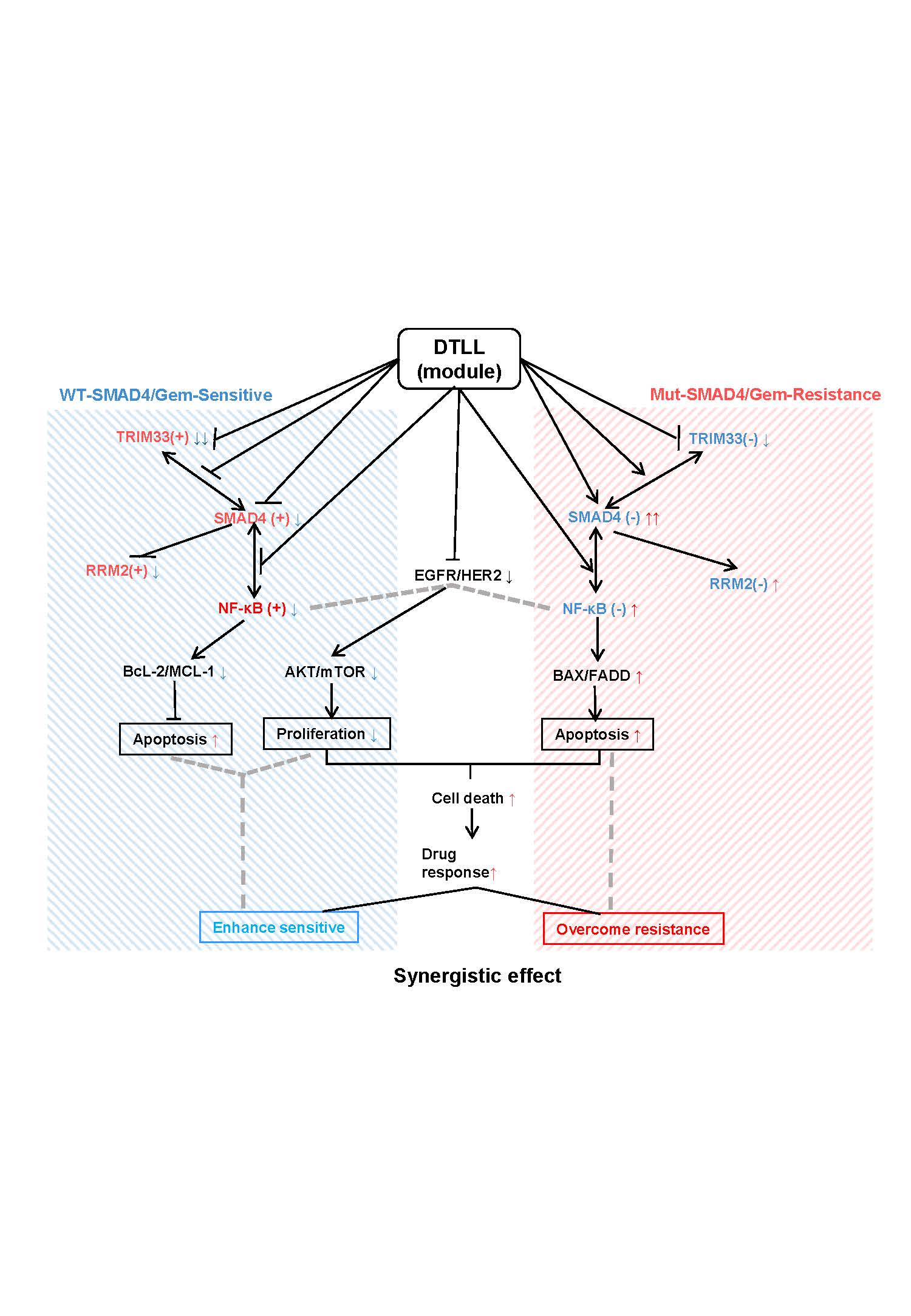
We have generated a highly potent bispecific antibody‒drug conjugate (ADC)-like agent, a dual-targeting EGFR/HER2 ligand-based lidamycin (DTLL). Our previous findings demonstrated that DTLL was superior to free LDM alone in ovarian carcinoma, PDAC, and esophageal cancer [13-15]. In our study, we aimed to evaluate DTLL efficacy when given in combination with gemcitabine by using gemcitabine-sensitive and gemcitabine-resistant PDAC models and to elucidate the relevant mechanisms in gemcitabine-resistant PDAC. Interestingly, DTLL is capable of sensitizing both gemcitabine-resistant and gemcitabine-sensitive cells to gemcitabine (or even other chemotherapeutic agents) through distinct action mechanisms according to different SMAD4 profiles. As shown in the schematic illustration model in Fig. 1, our mechanistic studies implied that DTLL combinational treatment might not only prevent neoplastic proliferation via blockage of ATK/mTOR signaling and antiapoptotic proteins (Bcl-2 and MCL1) mediated by impaired NF-kB function if given to SMAD4-sufficient/gemcitabine-sensitive PDAC cells but also restore the bioactivity of SMAD4 as a tumor suppressor to trigger its downstream NF-kB-regulated signaling of cell apoptosis (BAX/FADD) in SMAD4-deficient/gemcitabine-resistant tumors. The inhibitory effectiveness of DTLL alone or in combination with gemcitabine revealed that it functions as a double-edged sword in PDAC tumor growth, with totally different responsive behaviors and action mechanisms on synergistic efficacy between resistant and sensitive PDAC. We speculate that DTLL seems to act as a novel module of the SMAD4 driver that normalizes its function according to the protein level and genetic status in PDAC and then adjusts the NF-kB, TRIM33, gemcitabine-metabolic enzymes (such as RRM2) and proteomic profiles of PDAC cells, which explains the synergistic effect on tumor growth of DTLL in combination with gemcitabine.
Taken together, chemoresistance is a main limitation for the treatment of pancreatic ductal adenocarcinoma (PDAC). SMAD4 genetic status is responsible for SMAD4 protein levels, and its R100T mutation contributes to loss of SMAD4 protein and function with rapid protein degradation, leading to gemcitabine resistance in PDAC cells. Here, we show that DTLL overcomes gemcitabine resistance in PDAC preclinical models by mechanisms involving the tumor suppressor SMAD4. Our findings may reveal a potential combination therapy for overcoming resistance of PDAC mediated by SMAD4, which paves the way toward a long-sought tumor suppressor reactivation approach and lays the foundation for future precision medicine as the first step.
References
- Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016; 16: 553-565.
- Wang R, Cheng L, Xia J, Wang Z, Wu Q, Wang Z. Gemcitabine resistance is associated with epithelial-mesenchymal transition and induction of HIF-1alpha in pancreatic cancer cells. Curr Cancer Drug Targets. 2014; 14: 407-417.
- Kotteas E, Saif MW, Syrigos K. Immunotherapy for pancreatic cancer. J Cancer Res ClinOncol. 2016; 142: 1795-1805.
- Yamaoka T, Ohba M, Ohmori T. Molecular-targeted therapies for epidermal growth factor receptor and its resistance mechanisms. Int J Mol Sci. 2017; 18(11): 2420.
- Dreyer SB, Chang DK, Bailey P, Biankin AV. Pancreatic cancer genomes: implications for clinical management and therapeutic development. Clin Cancer Res. 2017; 23(7): 1638-1646.
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008; 321: 1801-1806.
- Kang YW, Lee JE, Jung KH, Son MK, Shin SM, Kim SJ, et al. KRAS targeting antibody synergizes anti-cancer activity of gemcitabine against pancreatic cancer. Cancer Lett. 2018;438: 174-186.
- Fiorini C, Cordani M, Padroni C, Blandino G, Di AgostinoS, Donadelli M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim Biophys Acta. 2015; 1853: 89-100.
- Wörmann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Görgülü K, et al. Loss of P53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016; 151:180-193.
- Cui Y, Brosnan JA, Blackford AL, Sur S, Hruban RH, Kinzler KW, et al. Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clin Cancer Res. 2012; 18:6519-6530.
- Nakajima E, Drezner N, Xiaoxue Li, Mishra-Kalyani P, Liu Y, Zhao H, Bi Y, et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin Cancer Res. 2022 Apr 14;28(8):1482-1486.
- Manji GA, Olive KP, Saenger YM, Oberstein P. Current and emerging therapies in metastatic pancreatic cancer. Clin Cancer Res. 2017; 23(7):1670-1678.
- Guo XF, Zhu XF, Shang Y, Zhang SH, Zhen YS. A bispecific enediyne-energized fusion protein containing ligand-based and antibody-based oligopeptides against epidermal growth factor receptor and human epidermal growth factor receptor 2 shows potent antitumor activity. Clin Cancer Res. 2010; 16: 2085-2094.
- Cao R, Song WP, Ye C, Liu XJ, Li L, Li Y, et al. Internal enhancement of DNA damage by a novel bispecific antibody-drug conjugate-like therapeutics via blockage of mTOR and PD-L1 signal pathways in pancreatic cancer. Cancer Med. 2019; 8(2): 643-655.
- Guo XF, Zhu XF, Yang WC, Zhang SH, Zhen YS. An EGFR/HER2-Bispecific and enediyne-energized fusion protein shows high efficacy against esophageal cancer. PLoS One; 2014; 9: e92986.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


![When PSMA-targeted therapy is not enough: high-risk localized prostate cancer after repeated [177Lu]Lu-PSMA radioligand therapy](/cdn-cgi/image/metadata=copyright,fit=scale-down,format=auto,quality=95,width=256,height=256/https://public-storage.zapnito.com/Ku6h7Yyp4Q0LXqRRMICCHR2v4LcOsmxMrmDPtOYuI1c)

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in