Early maturation and hyperexcitability is a shared phenotype of cortical neurons derived from different ASD-associated mutations
Published in Neuroscience

Autism spectrum disorder (ASD) is a neurodevelopmental disorder, where the patients often exhibit delayed development in motor and speech skills during infancy and varying degrees of difficulty in social interaction and communication later on in life. It is presented differently in each individual, ranging from mild to severe impairments. Approximately, one in 36 children in the US is affected by autism.
The risk for ASD is estimated to be influenced by genetic factors in the range of 10-20%.The interplay between gene variants and environmental factors, including parental age and birth complications, may play a significant role in determining an individual's susceptibility to this complex condition.
Numerous genes and genetic variants have been identified as causative factors for autism. These genetic abnormalities affect various aspects of cell communication and signal transmission, including genes involved in signal transmission and synapse function, among others. The wide range of mutations affects different genes and pathways.
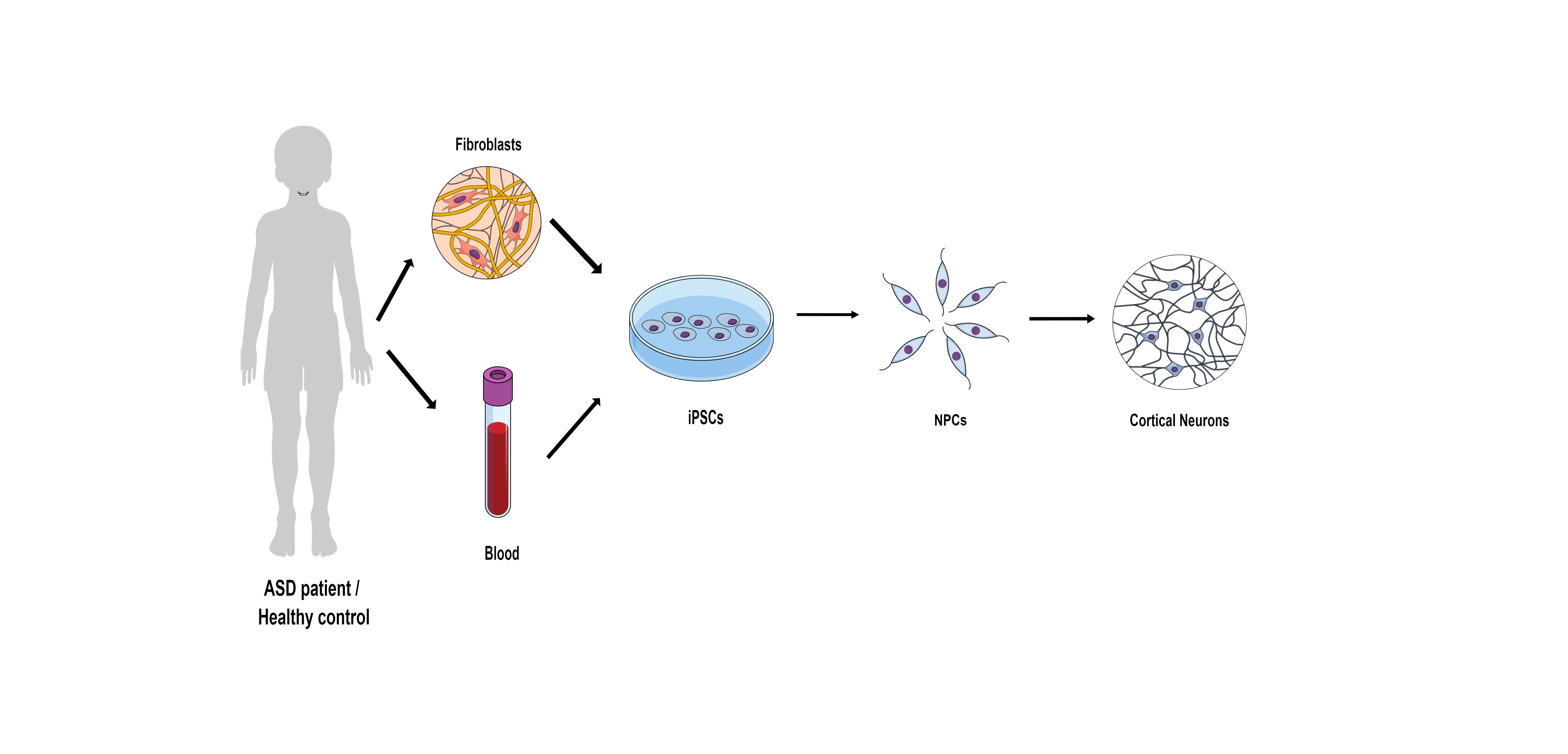
Some of the genetic-related ASD cases were modeled using mice and behavioral abnormalities were reported. At the cellular level, several genetic mutations have been shown to cause synaptic deficits in slice recordings. We were interested in studying human models using iPSC models. Importantly, the differentiation from iPSCs allowed us to follow the developmental stages that occur in utero or in the babies’ first months after birth. We were able to obtain blood samples from patients with four specific mutations: GRIN2B, SHANK3, UBTF, and a chromosomal duplication in the 7q11.23 region and first-degree relatives as controls.
The Dup7 mutation is a rare genetic syndrome resulting from a micro-duplication in section q11.23 of chromosome 7. This mutation has been associated with alterations in the ELN gene, which codes for the extracellular matrix protein elastin. These alterations are linked to connective tissue malformations observed in human patients. SHANK3 is a gene located at the terminal long arm of chromosome 22. It codes for a vital scaffolding protein found in various tissues, particularly in synapses of neurons in the brain. The GRIN2B mutation leads to the production of a nonfunctional GluN2B protein, an important component of NMDA receptors that play a role in neuronal impairments. UBTF is a gene responsible for coding a transcription factor critical for rRNA transcript synthesis in the nucleolus. Loss of UBF leads to nuclear disruptions, including inhibition of cell proliferation and cell death. These genes are not connected neither in their location nor in their function and therefore we did not expect to find common phenotypes. The studies were done independently because there did not seem to be any connection other than some shared symptoms of the patients.
In our study, we utilized peripheral blood mononuclear cells isolated from blood. These cells were reprogrammed into iPSCs using the Sendai reprogramming kit. Subsequently, these cells were differentiated into cortical neurons. The cortex is a region known to be involved and impaired in individuals with ASD.
To measure the developmental stages of the neurons, we started the experiments early – when the neurons were in their developmental stages. We were surprised to see that these different mutations shared similar abnormalities during their development. The observed changes in the four mutations included increased sodium currents, larger amplitudes and rate of excitatory postsynaptic currents (EPSCs), and an elevated number of evoked action potentials in response to current stimulation during early-stage cell development. These all indicate that the patients’ neurons have matured much faster than the neurons of the unaffected siblings. Additionally, we noted a reduction in GABA-positive cells and an increase in the excitatory-inhibitory ratio in these mutant neuronal cultures. It is interesting to note that these four mutations are also associated with epilepsy and the increase in excitability with the reduction in number of the GABA-ergic neurons may contribute to epilepsy in the patients.
In fact, in our previous study on the IQSEC2 mutation which is another ASD-associated mutation, we observed that mutant cells exhibited excessive excitability in the early phase and also a reduction in the number of GABA-ergic neurons. The IQSEC2 mutations are also highly associated with epilepsy. It is interesting to note that in the IQSEC2 study, we observed increased mortality as they aged, compared to healthy cells with normal growth. We have not yet measured the aged neurons in the four mutations in this study and it will be interesting to seek the increased mortality in these mutations as well.
Based on our findings, it is plausible that this early hyperexcitability occurs prenatally since the measured human neurons were differentiated for 4-5 weeks, indicating that they were not fully developed or matured. We speculate that there may be a connection between this early hyperexcitability and subsequent synaptic deficits, as this hyperexcitability could potentially be neurotoxic to the cells at such an early developmental stage. It is possible that this rapid development of the nerve cells ultimately causes damage to those nerve cells since these cells have not yet developed protective mechanisms. Future studies that we are working on in our laboratory are focusing on finding compounds and drugs that hopefully will slow down this rapid development and may rescue some of the phenotypes.
References
Brant, B., Stern, T., Shekhidem, H.A. et al. IQSEC2 mutation associated with epilepsy, intellectual disability, and autism results in hyperexcitability of patient-derived neurons and deficient synaptic transmission. Mol Psychiatry 26, 7498–7508 (2021).
Follow the Topic
-
Translational Psychiatry
This journal focuses on papers that directly study psychiatric disorders and bring new discovery into clinical practice.

Related Collections
With Collections, you can get published faster and increase your visibility.
Moving towards mechanism, causality and novel therapeutic interventions in translational psychiatry: focus on the microbiome-gut-brain axis
Publishing Model: Open Access
Deadline: Nov 15, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in