Electrochemical carbon−carbon coupling with enhanced activity and racemate stereoselectivity by microenvironment regulation
Published in Chemistry

Enzymes are characteristic of catalytic efficiency and specificity by maneuvering multiple components in concert at a confined nanoscale space, enabling high efficiency and catalytic specificity towards specific products1, 2. Inspired by the nature design, synthetic catalysts have been recently developed with sophisticated structure to employ noncovalent interactions with the reactants in a manner similar with an enzyme binding-pocket3, 4. However, achieving such configuration in artificial catalysts remains challenging.
Herein, we report a microenvironment regulation strategy by modifying carbon paper (CP) with hexadecyltrimethylammonium bromide (CTAB), delivering electrochemical carbon−carbon coupling of benzaldehyde with enhanced activity and racemate stereoselectivity. The CTAB-modified electrode–electrolyte interface creates an optimal microenvironment for electrocatalysis—it engenders dipolar interaction with reaction intermediate, giving 2.2-fold higher reaction rate (from 0.13 to 0.28 mmol h-1 cm-2); Moreover, it repels interfacial water and modulates the conformational specificity of reaction intermediate by facilitating intermolecular hydrogen bonding, affording 2.5-fold higher diastereomeric ratio of racemate to mesomer (from 0.73 to 1.82). We expect that the microenvironment regulation strategy will lead to advanced design of electrode–electrolyte interface for enhanced activity and (stereo)selectivity that mimics enzymes.
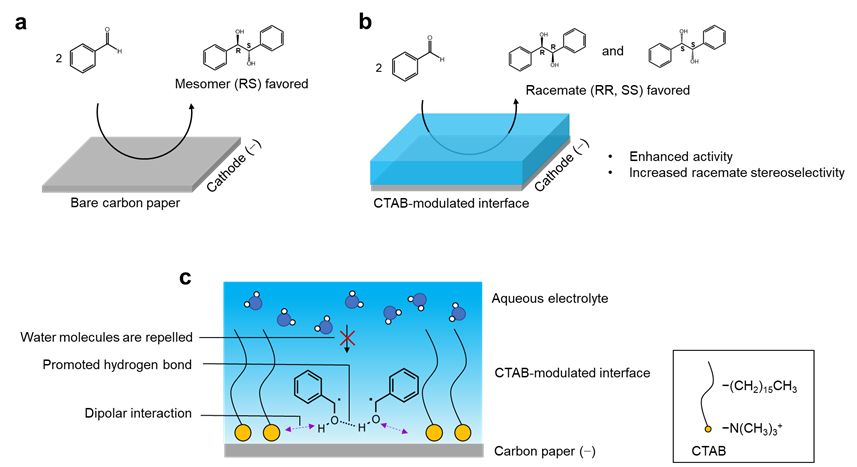
Fig. 1 | Microenvironment regulation for electrochemical pinacol coupling of benzaldehyde. Production distribution over a carbon paper and b CTAB-modified carbon paper. c Microenvironment at electrode–electrolyte interface of CTAB-modified carbon paper: confinement of ketyl radical via dipolar interaction with enhanced activity; modulation of conformational specificity of ketyl radical via repelling interfacial water and promoted hydrogen bond between ketyl radicals with enhanced racemate stereoselectivity.
To verify the ordered arrangement of CTAB molecules, we comprehensively adopted in-situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and electrochemical impedance spectroscopy (EIS) characterization5. The data consistently demonstrate that CTAB molecules undergo the conformational inversion at around -1.0 V versus Ag/AgCl and its hydrophobic tail shifts from facing to the electrode to facing to the electrolyte.
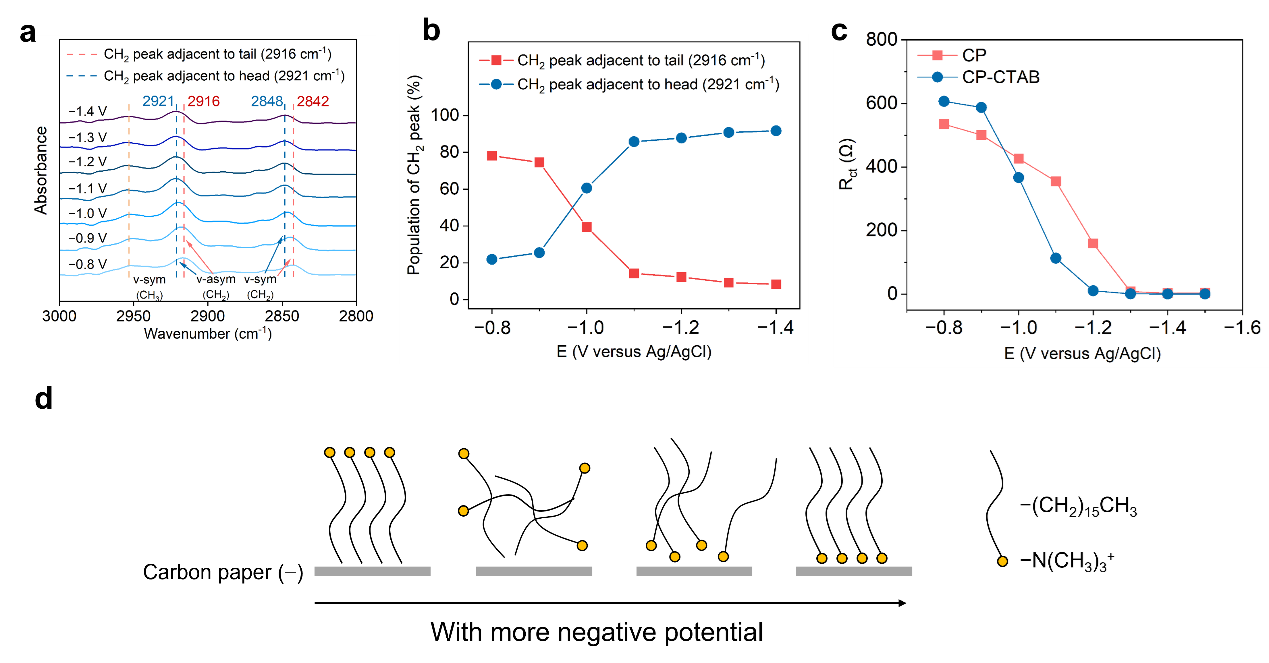
Fig. 2 | Adsorption and conformation of CTAB at electrode–electrolyte interface. a In-situ ATR-SEIRAS spectra at different potentials over CP-CTAB electrode in the range of 3000~2800 cm-1. b In-situ ATR-SEIRAS spectra, showing potential-dependent population of CH2 peaks in CTAB. c Resistance of charge transfer over CP and CP-CTAB electrodes at different potentials. d Schematic illustration of potential-dependent conformational transformation of CTAB molecules at electrode–electrolyte interface.
After rigorous electrochemical comparative experiments and in situ characterization, it is jointly demonstrated that the ordered arrangement of CTAB molecules regulates reaction activity and racemate stereoselectivity through dipolar and hydrophobic interactions, respectively. Based on the collective evidences, the plausible reaction mechanism is proposed. Part I: Without CTAB, benzaldehyde molecules orientate randomly and water layer is formed over CP interface. Part II: In the presence of CTAB, CTAB is adsorbed over CP interface using its head group under negative potential, leaving its long-chain facing to electrolyte with an ordered arrangement. The formed microenvironment enhances dipolar interaction between the head group of CTAB and benzaldehyde, and also repels interfacial water. Part III: At more negative potential (under electrochemical reaction condition), ketyl radical is generated as the reaction intermediate via a PCET process, and it is stabilized by CTAB via dipolar interaction. Meanwhile, the hydrogen bonds between two ketyl radicals are more prominent due to fewer interfacial water molecules at electrode–electrolyte interface. Part IV: As a result of dipolar interaction and interfacial water repelling, the presence of CTAB enhances the reaction rate, and producing racemates is more favorable as the C−C radical coupling products.
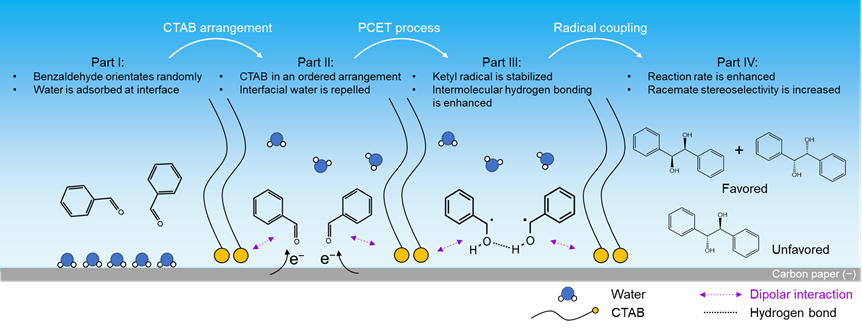
Fig. 3 | Proposed reaction mechanism of electrochemical benzaldehyde C−C coupling reaction. The reaction process is depicted in four parts (from left to right). Part I: without CTAB, reactant orientates randomly and water layer is formed over CP surface. Part II: CTAB is adsorbed over CP interface in an ordered arrangement, which enhances dipolar interaction with reactant and repels interfacial water. Part III: at more negative potential, ketyl radical is generated via a PCET process, and it is stabilized by CTAB at CP surface via dipolar interaction. Moreover, the conformation of ketyl radical is modulated by repelling away interfacial water molecules, with enhanced intramolecular hydrogen bonding. Part IV: Radical coupling takes place with enhanced activity and racemate stereoselectivity.
We recognize mesomer stereoselectivity was not fully suppressed by CTAB modification, which is not unreasonable because controlling the conformation of transient ketyl radicals remains challenging. Meanwhile, the relatively weak binding energy of hydrogen bond (typically ~20 kJ/mol)6 suggests that either the interaction between hydroxyl groups (that favors racemate) or between hydroxyl group and water (that favors mesomer) may not be strong enough to prevail. More studies can be carried out in the future to design a microenvironment with stronger interaction and spatially more optimized configuration with the reactant and/or intermediates, which we anticipate would be efficient to further enhance the stereoselectivity.
References
1. Cobb, S.J. et al. Fast CO(2) hydration kinetics impair heterogeneous but improve enzymatic CO(2) reduction catalysis. Nat. Chem. 14, 417-424 (2022).
2. Lee, C.C. et al. Evidence of substrate binding and product release via belt-sulfur mobilization of the nitrogenase cofactor. Nat. Catal. 5, 443-454 (2022).
3. Li, Z. et al. Electrocatalytic synthesis of adipic acid coupled with H2 production enhanced by a ligand modification strategy. Nat. Commun. 13, 5009 (2022).
4. Wakerley, D. et al. Bio-inspired hydrophobicity promotes CO(2) reduction on a Cu surface. Nat. Mater. 18, 1222-1227 (2019).
5. Ge, W. et al. Dynamically Formed Surfactant Assembly at the Electrified Electrode-Electrolyte Interface Boosting CO2 Electroreduction. J. Am. Chem. Soc. 144, 6613-6622 (2022).
6. Bjorneholm, O. et al. Water at Interfaces. Chem. Rev. 116, 7698-7726 (2016).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in