Enabling late-stage drug diversification by high-throughput experimentation with geometric deep learning
Published in Chemistry
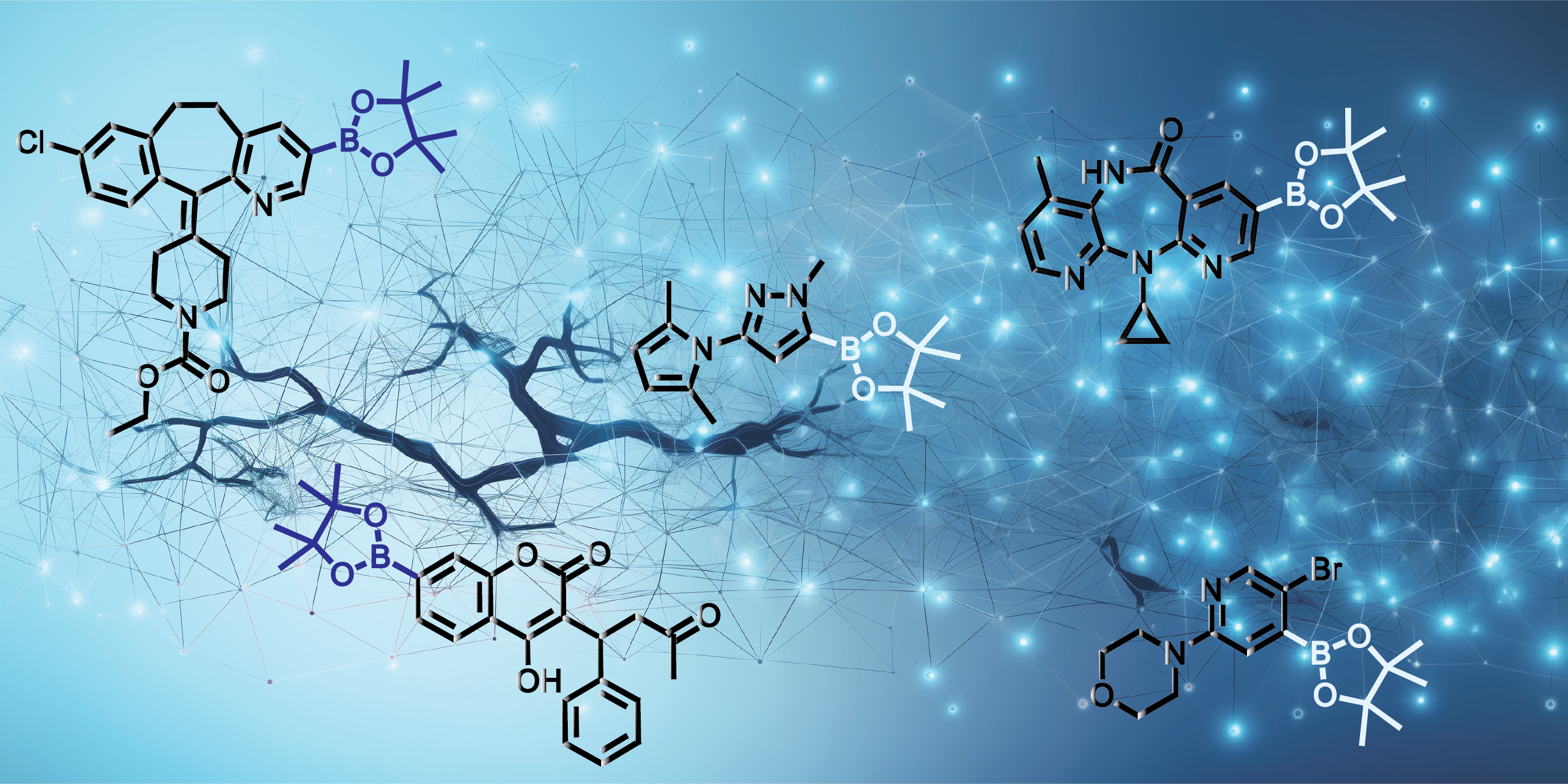
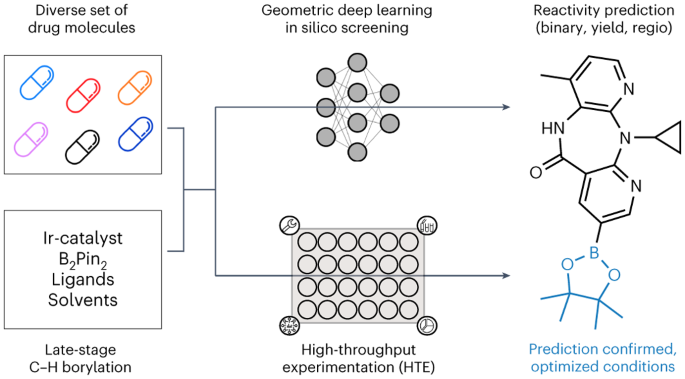
Late-Stage Functionalization with C-H Borylation
Late-stage functionalization (LSF) is an appealing approach to enhance the properties of drug candidates, as it enables the modification of organic scaffolds and the exploration of structure-activity relationships (SARs) in medicinal chemistry [1]. Nevertheless, the chemical complexity of drug molecules, characterized by a wide range of functional groups, C-H bonds, and site-specific requirements, poses an obstacle when it comes to achieving customized late-stage diversification. Among the various methods for LSF, C-H borylation is recognized as one of the most versatile techniques for quickly diversifying compounds, providing a sturdy foundation for subsequent C-C bond connections [2]. Despite its considerable potential, late-stage functionalization (LSF) in drug discovery is currently underutilized, and the reported applications are limited, often concentrated on specific types of reactions. The challenge lies in establishing general principles for predicting reactivity and selectivity across the wide array of C-H activation reactions, which is complicated due to the diverse nature of the substrates involved. As a result, LSF frequently depends on resource-intensive experimentation, a method that is incompatible with the constraints often encountered in medicinal chemistry projects.
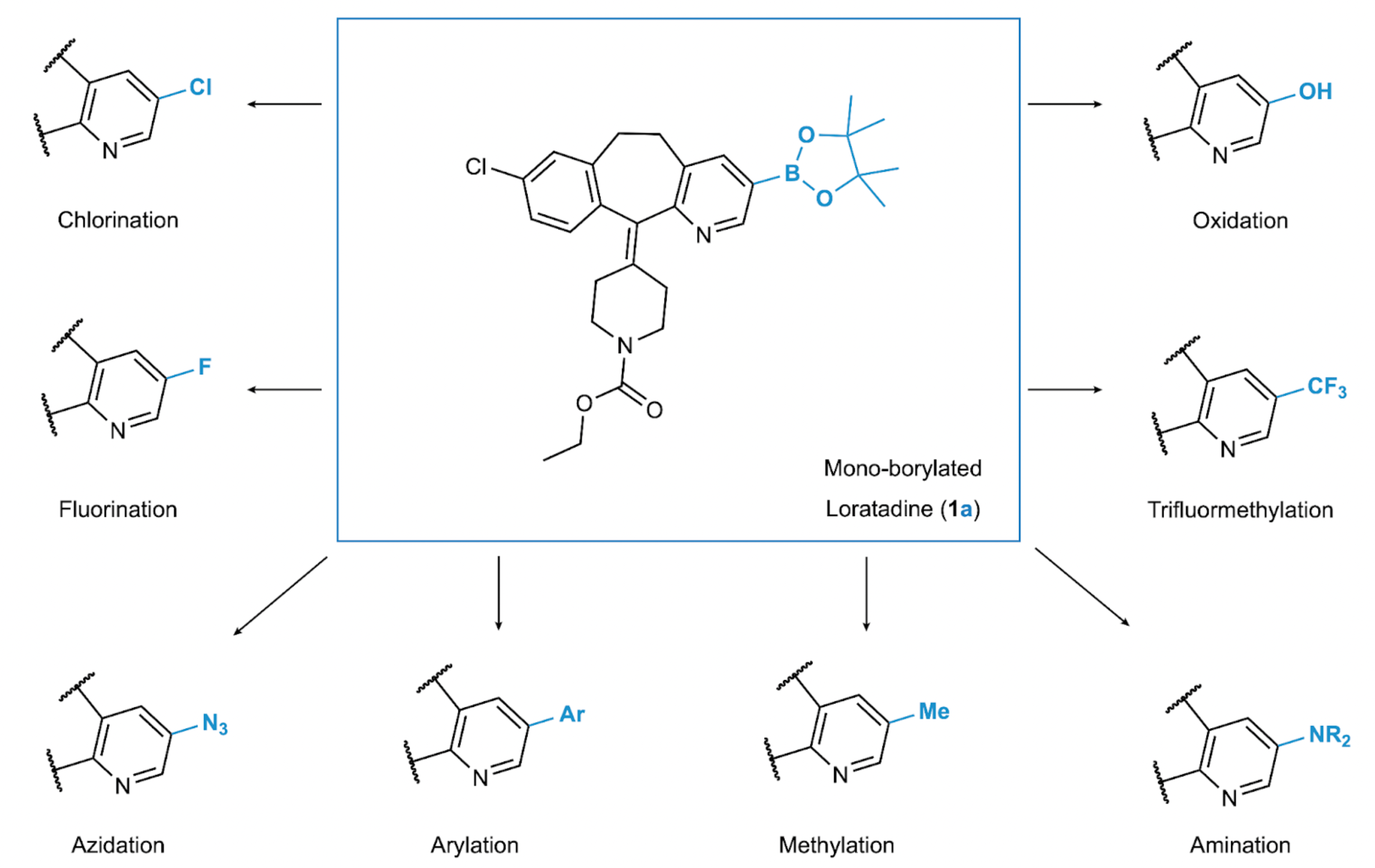
Fig. 1. Late-stage borylation of a drug molecule. The example showcases mono-borylated Loratadine (1a), which can be obtained by borylating the drug Loratadine (1) and then further modified to incorporate various functional groups.
High Throughput Experimentation for Data Generation
To overcome the data limitations associated with C-H borylation reactions on complex drug molecules, we implemented a high-throughput experimentation (HTE) platform that employs semi-automated, miniaturized, and low-volume screening methods. This HTE setup allowed for the swift and consistent testing of multiple reactions simultaneously, facilitating the screening of a wide range of substrates and reaction conditions within a matter of weeks. In total, we conducted screenings on 23 diverse commercial drug molecules and a subset of 17 frequently encountered drug-like fragments. This effort resulted in the generation of a comprehensive dataset encompassing 956 reactions. When combined with reactions previously documented in the literature, covering smaller substrates, we were able to assemble a dataset of 2,257 C-H borylation reactions [3].
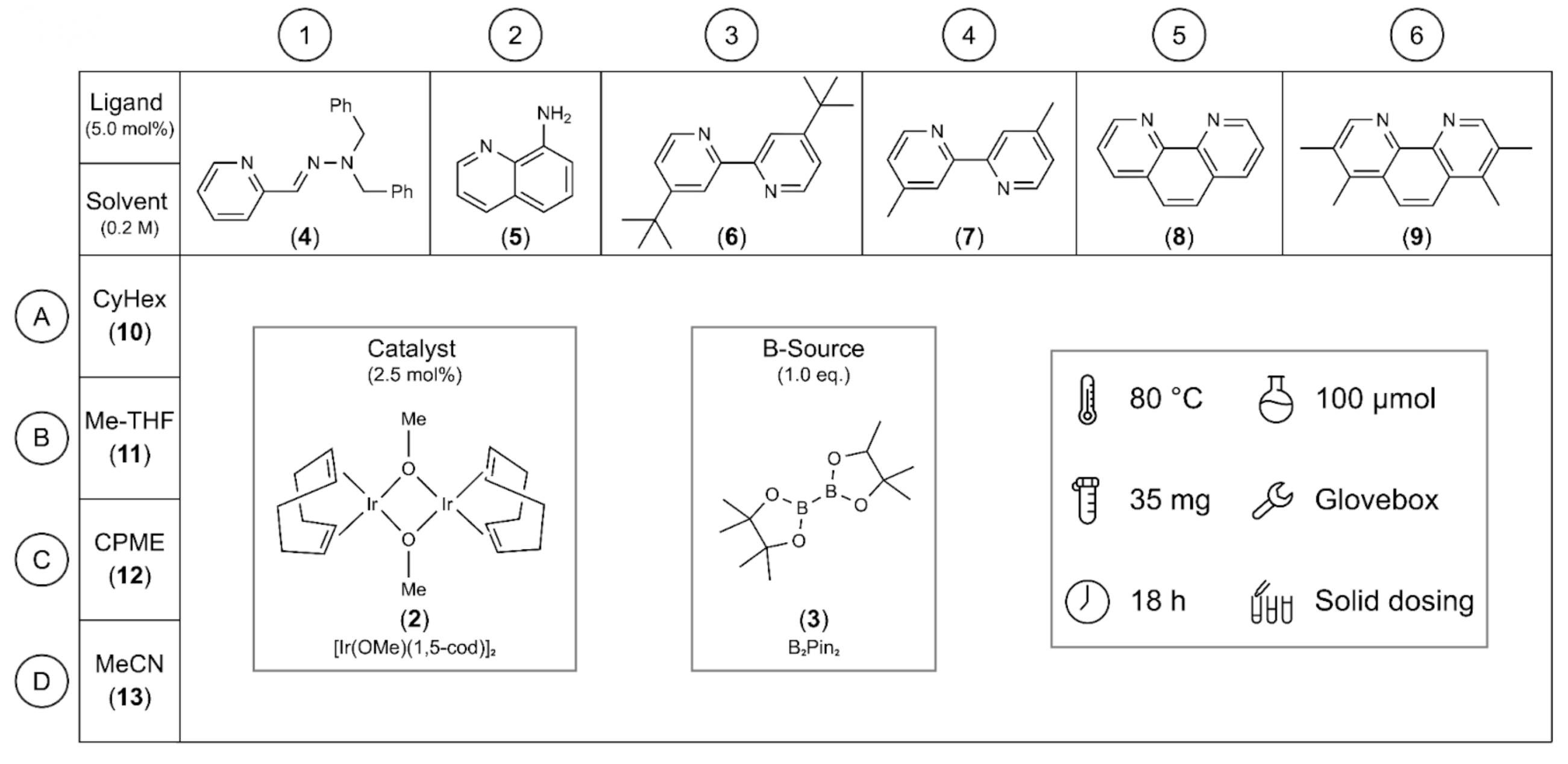
Fig. 2. Schematic of the 24-well borylation screening plates that were used in the experiments. We conducted screening tests involving one catalyst (2), one boron source (3), six different ligands (4-9), and four distinct solvents (10-13) for all the starting materials.
Graph Machine Learning Enables Reaction Prediction
Graph neural networks have found extensive applications in tasks related to molecular feature extraction and property prediction [4,5]. In our research, we introduced customized Graph Transformer Neural Networks (GTNNs) designed for predicting binary reaction outcomes, reaction yields, and regioselectivity. GTNNs were specifically engineered to seamlessly incorporate both three-dimensional (3D) and electronic features, while also accounting for reaction conditions relevant to these different tasks. Our results demonstrate that the developed GTNNs outperform baseline models in predicting reaction yields, achieving a mean absolute error of 4.2% [3].
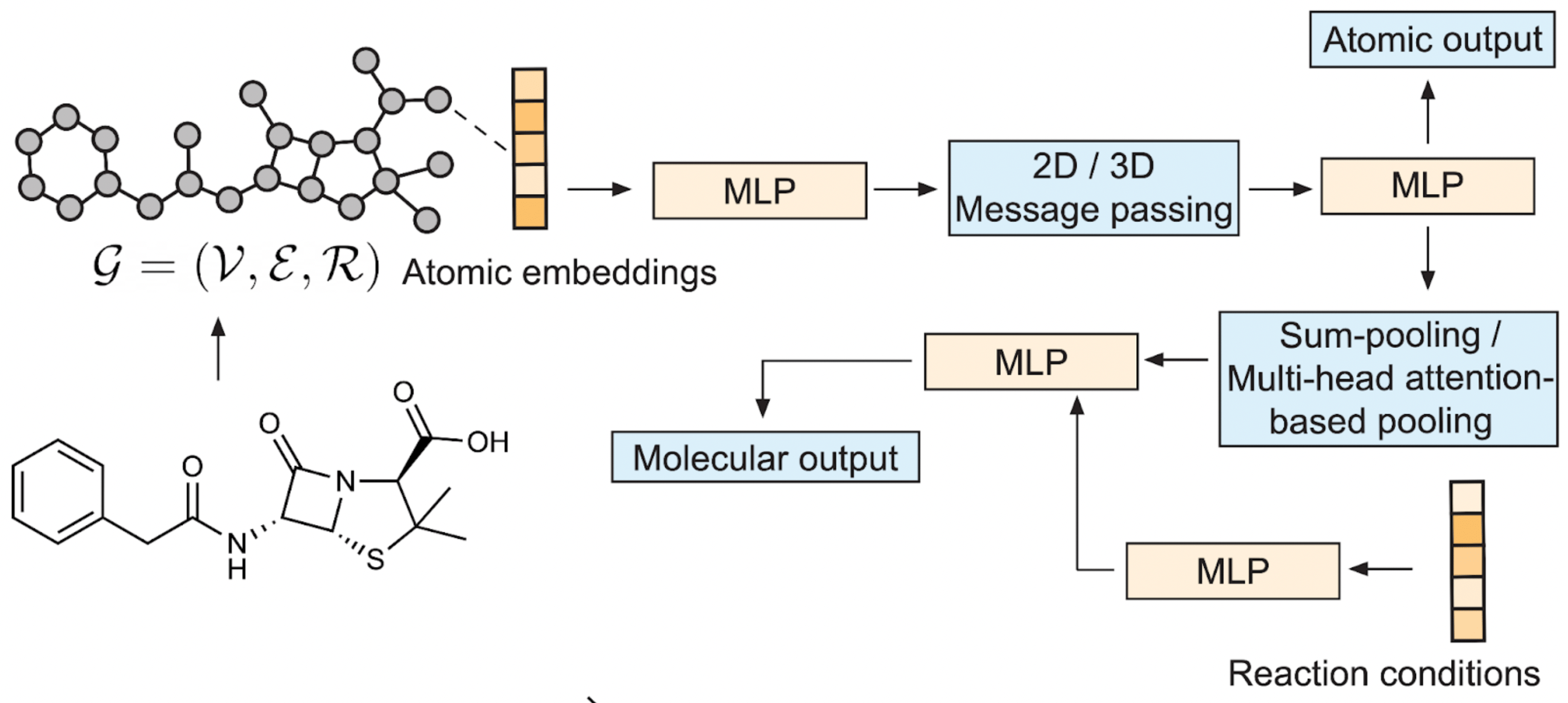
Fig. 3. Schematic of the graph neural network architecture. After passing the atomic features through the first MLP, the atomic features are updated via three message-passing layers. Subsequently, the learned atomic features are either transformed directly to the regioselectivity output, or pooled to obtain a whole-molecule feature space. This learned molecular feature space is then combined with the embedded features of the reaction conditions and transformed into the reaction output via a final MLP.
Prospective Validation of Regioselectivity Prediction
A critical challenge encountered by medicinal chemists when developing C-H borylation reactions for new substrates is the issue of regioselectivity, which involves predicting which C-H bond in an organic molecule will undergo a desired transformation. Our research demonstrates that GTNNs, once trained, can accurately forecast regioselectivity for substrates that fall outside the established distribution. They are also effective at handling various substitution patterns, encompassing diverse electronic effects and steric hindrances. Through prospective validation of the trained GTNNs on six novel substrates, including three drugs and three fragments, our findings indicate that the model correctly predicted five out of seven experimentally observed borylation sites [3].
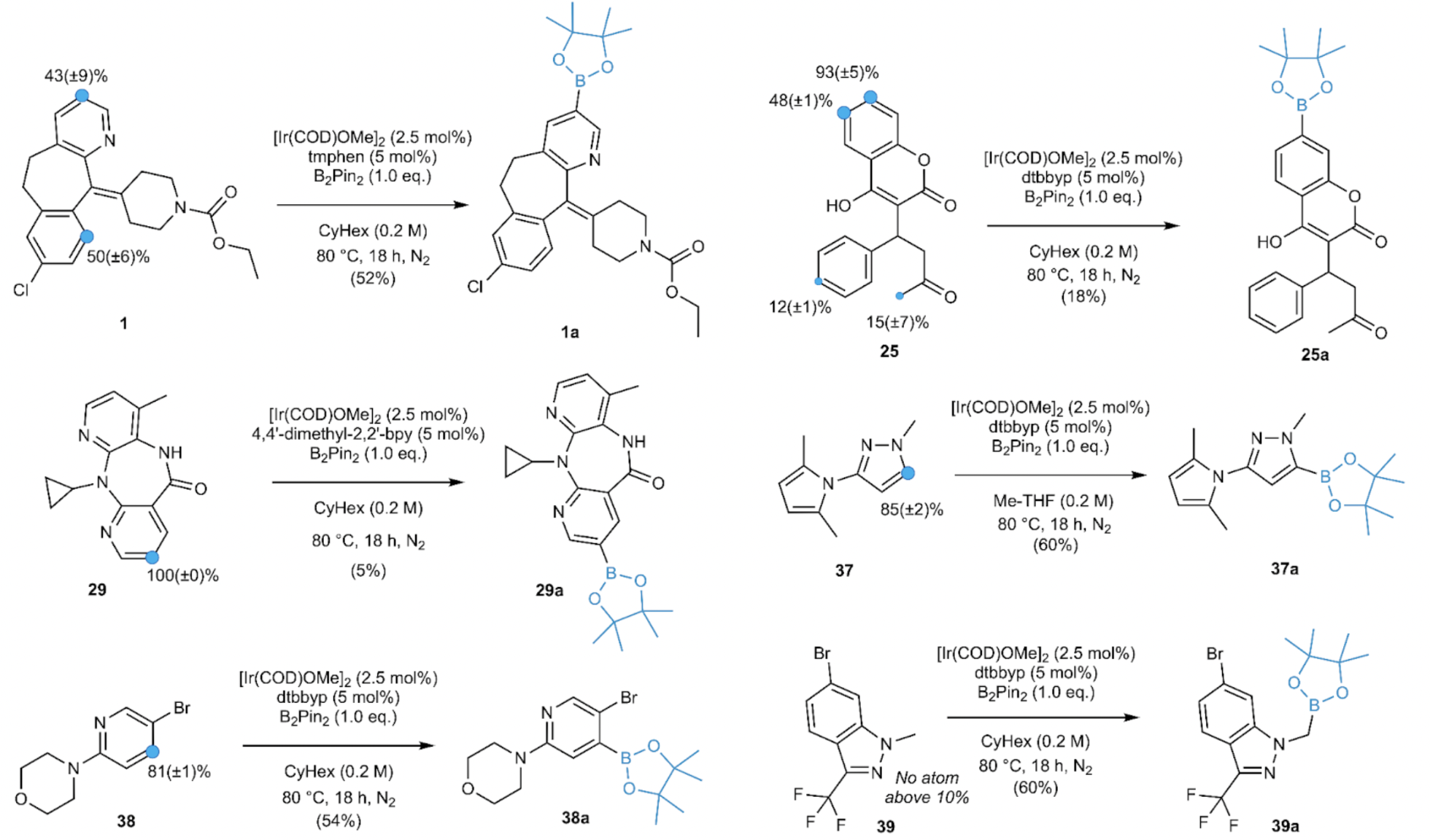
Fig. 4. Prospective experimental validation of regioselectivity prediction models that were trained on the literature data set. Validation is shown for three drugs, Loratadine (1), Warfarin (25), and Nevirapine (29), and three fragments, (37-39).
Simple User-Friendly Reaction Format
This study's success rested on the introduction of the implementation of a comprehensive, straightforward, and user-friendly reaction format known as SURF (Simple, Unified, and Readable Format). SURF serves as a standardized means of documenting reaction data using a structured tabular format, which only requires a basic understanding of spreadsheet applications. This format empowers chemists to record the synthesis of molecules in a manner that is both comprehensible to humans and machine-readable, facilitating easy sharing and seamless integration into machine-learning pipelines. SURF not only streamlined the experimental process but also heightened the reproducibility of reactions while rendering the data more readily accessible for future research and practical applications [3].
Reaction Predictions Hold the Potential to Impact Drug Discovery
The findings presented in this paper have profound implications for the field of drug discovery. The combination of LSF, HTE, and graph machine learning introduces new possibilities for efficiently and cost-effectively diversifying and optimizing drug candidates. Accurate prediction of reaction yields under different conditions and regioselectivity reduces the need for time-consuming and resource-intensive experimental work. This approach has been successfully applied to various projects, particularly in exploring borylation opportunities within drug discovery initiatives at F. Hoffmann La-Roche. In summary, the integration of reaction prediction and laboratory automation in drug discovery projects represents a pivotal step towards expediting drug development, reducing costs, and fostering a data-driven approach. This, in turn, holds the potential to contribute to the creation of safer and more effective medications for a wide range of medical conditions.
References
- Nippa, D. F. et al. Late-stage functionalization and its impact on modern drug discovery: Medicinal chemistry and chemical biology highlights. Chimia 76, 258–258 (2022).
- Hartwig, J. F. Borylation and silylation of C-H bonds: A platform for diverse C-H bond functionalizations, Acc. Chem. Res. 45, 864–873 (2012).
- Nippa, D. F., Atz, K. et al. Enabling late-stage drug diversification by high-throughput experimentation with geometric deep learning, Nat. Chem. (2023).
- Atz, K., Grisoni, F. & Schneider, G. Geometric deep learning on molecular representations, Nat. Mach. Intell., 3, 1023–1032 (2021).
- Isert, C., Atz, K. & Schneider, G. Structure-based drug design with geometric deep learning, Curr. Opin. Struct. Biol. 79, 102548 (2023).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in