Engineered cytokine receptors improve CAR T cell therapy for solid tumors
Published in Bioengineering & Biotechnology

While chimeric antigen receptor (CAR) T cell therapy has demonstrated impressive responses in hematologic malignancies, leading to FDA approval of six CAR T cell products for leukemia, lymphoma, and multiple myeloma, it has so far been less effective for the treatment of solid tumors and brain tumors. While several roadblocks are thought to impair CAR T cell activity in the solid tumor microenvironment (TME), we chose to address the limited availability of T cell-supporting cytokines due to previous preclinical and clinical observations that highlight the central role of cytokine signaling in effective T cell responses. In fact, cytokine availability is so essential to CAR T cell efficacy that treatment often incorporates lymphodepleting chemotherapy to increase the concentration of cytokines in the blood.
Many approaches have been investigated to provide cytokine support to T cells for the treatment of cancer, however these approaches have been limited by one or multiple factors. Cytokines act on many different cell types in the body, leading to toxicity if they are injected into the circulation or if T cells are modified to express high levels of certain cytokines. Additionally, the local concentration of cytokines in the tumor may be insufficient or T cells may downregulate the cognate cytokine receptor as they become activated, differentiate, or become exhausted in the TME. Therefore, we chose to engineer synthetic versions of cytokine receptors that would continuously provide cytokine support to CAR T cells without stimulating other cell types.
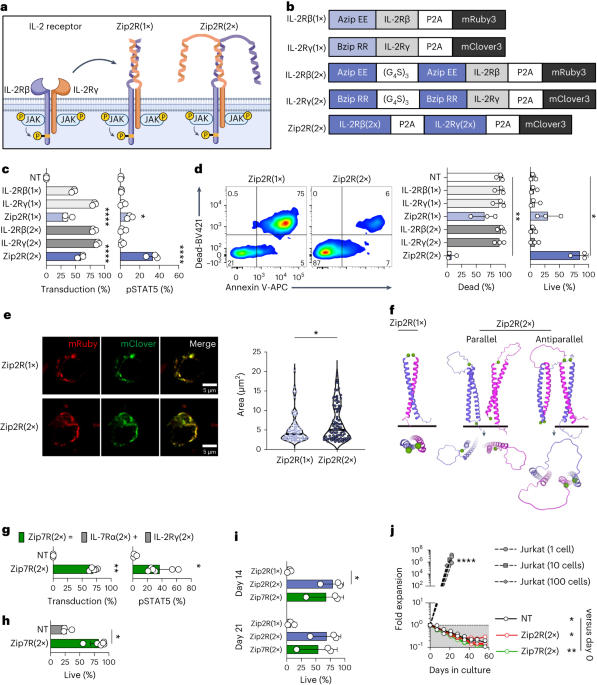
Natural cytokine signaling in T cells requires the interaction of an extracellular molecule and one or more receptor chains on the cell surface which couples ligand sensing with signal transduction. Since any of these components can be limiting, we reasoned that engineering a synthetic receptor that is always expressed and always signaling could provide a better and more targeted CAR T cell stimulus compared to previous approaches. We therefore replaced the extracellular domains of the interleukin (IL)-2 receptor beta and gamma chains with leucine zipper motifs to generate leucine zipper-based cytokine receptors (ZipRs). These leucine zippers were designed to form high affinity heterodimers, which in our system would hopefully induce IL-2 receptor dimerization and signaling without the need for cytokine binding. To our surprise, more than one pair of leucine zippers was required for robust activation of the signaling pathway, which could be explained by the formation of large order signaling complexes and multiple possible orientations of interaction that are not possible with only one pair.
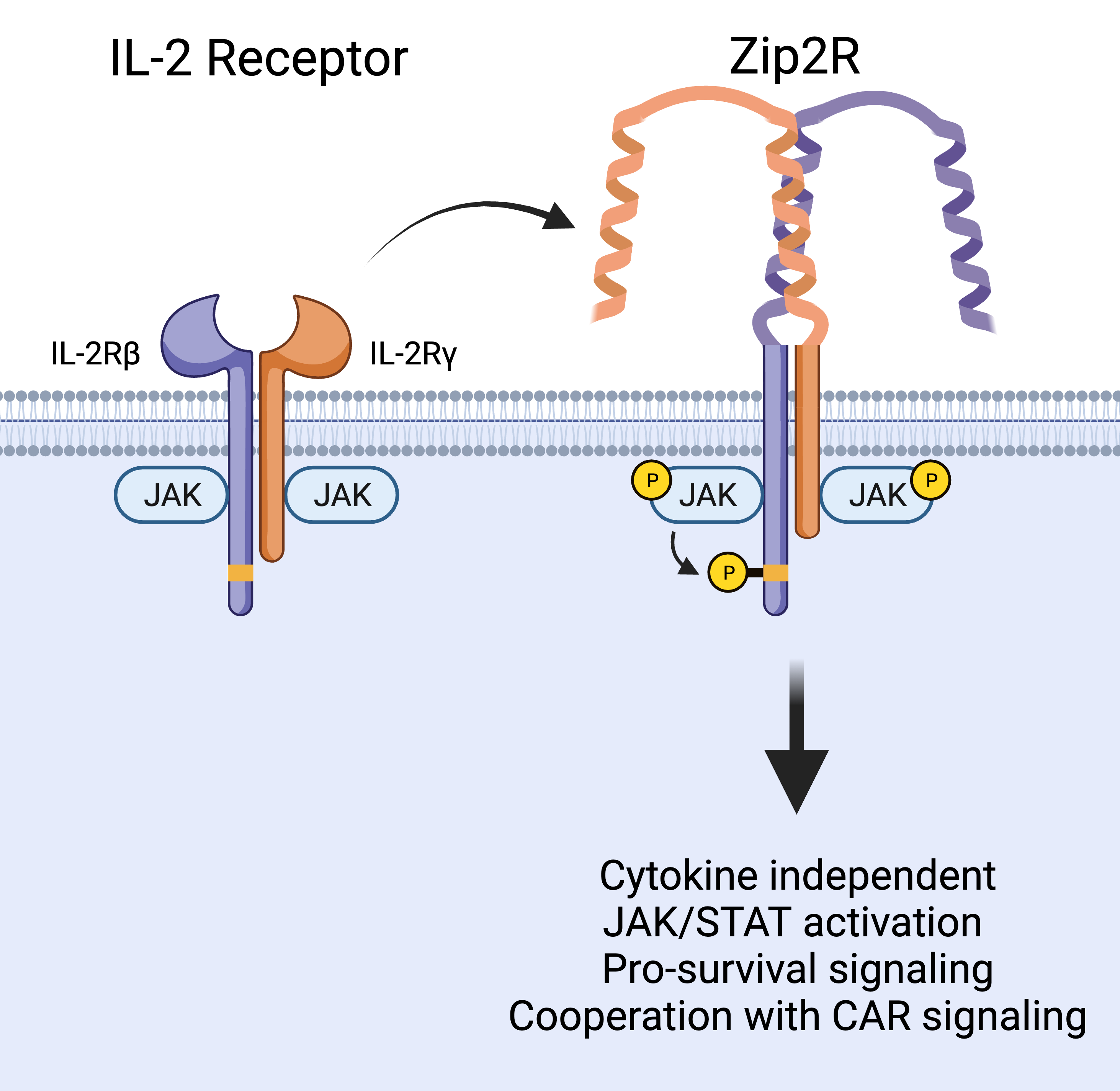
In contrast to physiological IL-2 signaling which promotes T cell survival and proliferation, our engineered leucine zipper-based IL-2 receptor (Zip2R) provided a survival signal to T cells without inducing proliferation. This is an important feature as it could allow CAR T cells to survive in circulation while migrating to the tumor, without causing uncontrolled growth. Instead, in vitro experiments demonstrated that Zip2R improved CAR T cell proliferation only after encountering antigen positive tumor cells. This cooperation with CAR signaling allowed CAR T cells to repetitively kill tumor tumors and expand, another important aspect of the system as an effective antitumor response requires intratumoral CAR T cells, which are outnumbered by tumor cells, to kill several times in order to clear the solid tumor mass. In fact, in a disseminated lung tumor model in which a low dose of CAR T cells was unable to control tumor growth, Zip2R expression rescued CAR T cell efficacy and led to tumor clearance in several mice.
With these encouraging results, we next extended our findings by designing variations of Zip2R. Multiple cytokine receptors complexes share individual receptor chains, such as the IL-2 receptor gamma chain that is shared by the receptors for IL-2, IL-7, and IL-21, or the IL-10 receptor beta chain which is shared by receptors for IL-10 and IL-22. As proof of the modularity of the ZipR system, we were able to show that individual receptor components can be swapped to tailor signaling to other cytokine pathways and in total demonstrated functionality for 6 different ZipRs. Similar to Zip2R, ZipRs designed from IL-7, IL-21, or IL-12 receptors were able to improve the antitumor activity of CAR T cells by activating diverse signaling pathways which cooperated with CAR-mediated signaling to boost their ability to kill tumor cells, allowing previously ineffective doses of CAR T cells to better respond to solid tumors in vivo. Finally, a deeper exploration of the transcriptomic and phosphoproteomic changes induced by Zip2R, Zip7R, or Zip12R suggested that diverse signaling pathways can be activated to improve CAR T cells and that analyses focusing only on JAK/STAT signaling may be insufficient as these receptors induced signaling associated with a variety of cellular processes including cell survival and proliferation, RNA splicing, chromatin remodeling, cell adhesion, cytoskeletal regulation, and metabolism. While we saw major improvements in CAR T cell antitumor activity with the different ZipRs, this system was not completely without risks. We observed toxicity from Zip7R CAR T cells in our lung tumor model, highlighting that some anatomical locations are more sensitive to CAR T cell therapy and that second genetic modifications to improve CAR T cell function will likely require the incorporation of safety features.
In these proof-of-concept studies, we demonstrated that ZipRs improve the ability of CAR T cells to treat solid tumors. Future work will focus on extending these findings to other tumor models, cytokine signaling pathways, and immune effector cells. Because of the unique mode of signaling provided by ZipRs, this approach also opens avenues of treatment that were previously unexplored or were discarded due to the non-specificity of cytokine signaling and we hope this will yield cures for patients with solid tumors.
Follow the Topic
-
Nature Biomedical Engineering
This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.

Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Medical Ultrasound: Emerging Techniques and Applications
Publishing Model: Hybrid
Deadline: Jan 29, 2027

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in