“Etching” Genomic Sequencing Reads to Develop the Fastest Structural Variation Caller
Published in Bioengineering & Biotechnology
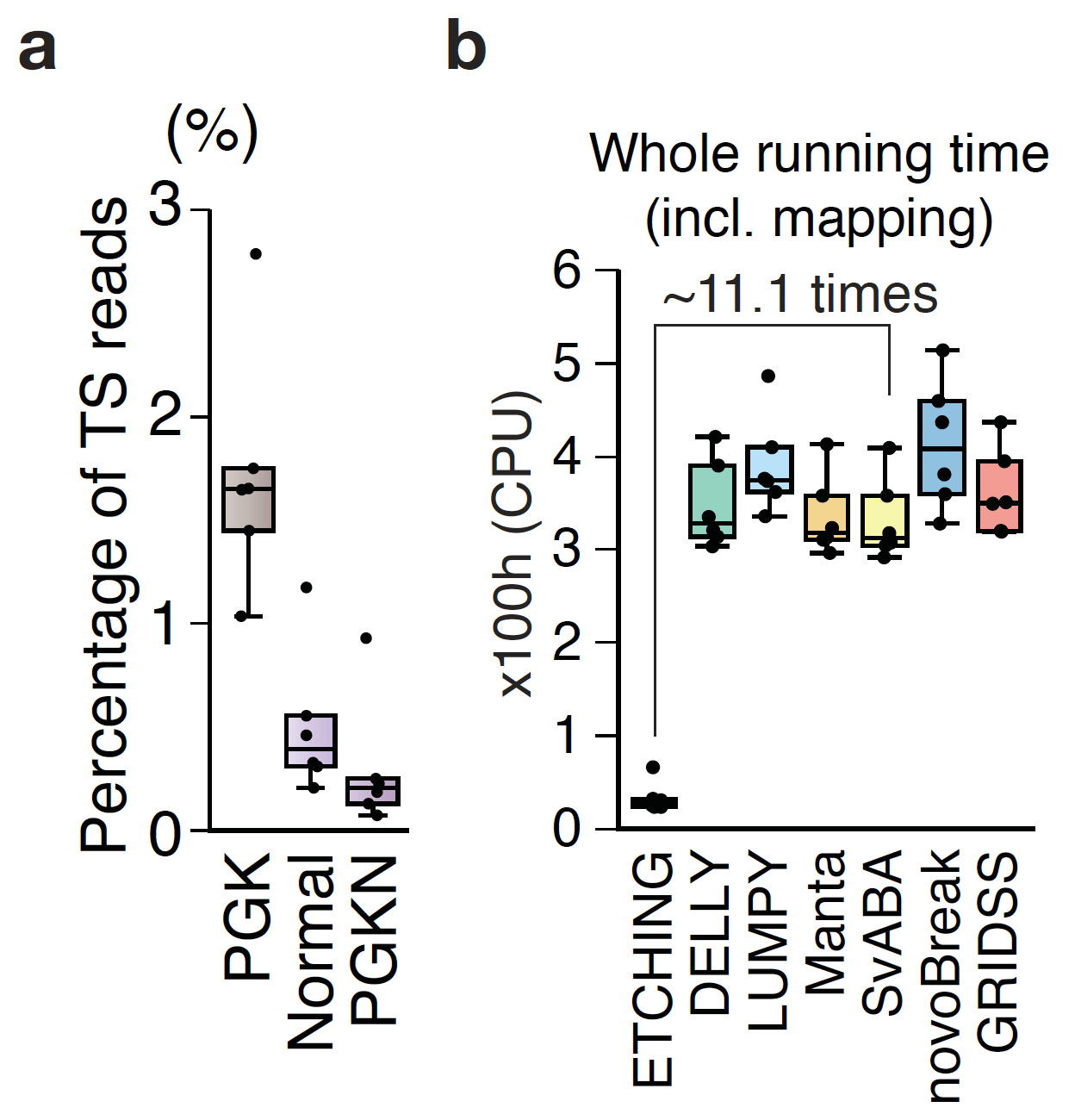

Etching genomic sequencing reads: Cancer diagnostics increasingly involve analysis of changes in tumor DNA, including structural variations (SVs) such as insertions, deletions, inversions, and fusions between genes. Detection of SVs from whole genome sequencing (WGS) data comes with very high computing costs because hundreds of millions of sequencing reads must be processed and mapped to the reference genome. Even worse, approximately half of the reads, mostly from repeat elements, are ambiguously mapped to multiple genomic loci, often causing the production of false positives and the veiling of true positives. The ability to collect only reads embedding mutational information would greatly reduce background noise as well as computing costs. Our recent study used a parallel matching algorithm to identify k-mers (that is, short sequences of length k) that were perfectly matched to the reference genome, leaving behind less than 1% of the reads, which are tumor-specific and embed somatic variations (Figure 1a) [1]. We refer to this method as “etching” because of the sharp drop in the number of sequencing reads when only those containing tumor-specific SVs remain, reminiscent of chemical etching. This process allowed us to robustly detect SVs with high specificity, sensitivity, and speed. In fact, our software, ETCHING (https://github.com/ETCHING-team/ETCHING), achieved results at least 11 times faster than the previous fastest tool with no compromise in performance (Figure 1b).

(TS, tumor-specific)
Democracy in genomics: Although the governments of some developed countries and consortium-based WGS projects have invested billions of dollars to collect millions of WGS datasets, managing population-scale WGS data has only been possible for tech giants, such as Google, Facebook, Microsoft, and Amazon, and a few top institutes with giant computing facilities [2-3]. A few companies and institutes have started to monopolize human genome research because of their ability to harness economies of scale. However, many small laboratories, including my own (BIGLab: http://big.hanyang.ac.kr), also wanted to join this exciting field and rub shoulders with the giant competitors. I believe that ETCHING, which requires only small computing resources, can democratize genomic research. In fact, the BIGLab runs a high performing computing (HPC) facility with several hundred processors, which provide the capacity (using common SV detectors) to process hundreds of WGS datasets per month. With ETCHING, however, our HPC facility can process thousands of WGS datasets in the same time period. I hope that, like us, many other small laboratories will start to use ETCHING to analyze big WGS datasets at the population level.
Detection of somatic variations with matched normal samples: Recent studies have reported that germline SVs are more prevalent in normal populations than we anticipated. An average person has tens of thousands of genomic SVs compared to the reference genome [4-5]. In contrast, pan-cancer studies have reported that tens to several hundreds of somatic SVs are present in a tumor sample [6]. To precisely define somatic SVs in tumor samples, a sample of healthy tissue from the same individual is essential. A majority of somatic SV callers take advantage of such matched normal samples to filter germline SVs that are also present in the normal sample. However, matched normal samples are not always available in clinical cancer sequencing studies due to various reasons including institutional policies, the cost of sequencing, issues related to consent, the amount of available sample, and so on [7]. Hence, the ability to predict druggable somatic SV targets with confidence without a matched normal sample will benefit not only clinicians but also patients. Notably, the performance of ETCHING using only tumor sample data was comparable with that of other SV callers using both tumor and matched normal samples for detecting somatic SVs from WGS data (Figure 2a) and cancer panel data (Figure 2b). Fast matching billions of k-mers derived from hundreds of normal genomes to tumor reads helped us to filter out not only prevalent germline variations but also sporadic variations.

Whole genome and cancer panel sequencing data: When this study was started by the Korea Post-Genome Project five years ago, the original goal was to develop a fast, high-performing somatic SV caller for WGS data. However, most domestic hospitals had started to sequence targeted cancer panels for cancer diagnoses because of the high cost of obtaining WGS data and the policies of health insurance companies, although the cost of WGS sequencing has continuously declined. Thus, we decided to develop a tool that could be applied to both WGS data and cancer panels. Fortunately, ETCHING robustly performed SV predictions and outperformed other tools for SVs with a low allele frequency (Figure 3), which led a genomics company to implement it as a module in a clinical pipeline for detecting SVs from liquid biopsy samples.

References
[1] Sohn et al., Ultrafast prediction of somatic structural variations by filtering out reads matched to pan-genome k-mer sets, Nature Biomed. Eng., In press.
[2] Goldfeder et al., Human genome sequenicng at the population scale: a primer on high-throughput DNA sequencing and analysis, Am J Eidemiol., 186(8):1000-1009, 2017.
[3] Tanjo et al., Practical guide for managing large-scale human genome data in research, Journal of Human Genetics, 66()39-52, 2020.
[4] Chaisson et al., Resolving the complexity of the human genome using single-molecule sequencing, Nature, 517(7536):608-11. 2015.
[5] Sudmant et al., An integrated map of structural variations in 2,504 human genomes, Nature, 526(7571):75-81. 2015
[6] Li et al., Patterns of somatic structural variations in human cancer genomes, Nature, 578(7793):112-121, 2020.
[7] Sun et al., A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal, PLoS Comput Biol 14(2):e1005965, 2018.
Follow the Topic
-
Nature Biomedical Engineering
This journal aspires to become the most prominent publishing venue in biomedical engineering by bringing together the most important advances in the discipline, enhancing their visibility, and providing overviews of the state of the art in each field.

Related Collections
With Collections, you can get published faster and increase your visibility.
Implantable wireless communication technologies
Publishing Model: Hybrid
Deadline: Nov 28, 2026
Medical Ultrasound: Emerging Techniques and Applications
Publishing Model: Hybrid
Deadline: Jan 29, 2027




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in