Faster, cheaper, better: FLASH-seq for full-length single-cell RNA-sequencing
Published in Bioengineering & Biotechnology

Explore the Research
Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq - Nature Biotechnology
FLASH-seq speeds up high-sensitivity scRNA-seq while sparing resources.
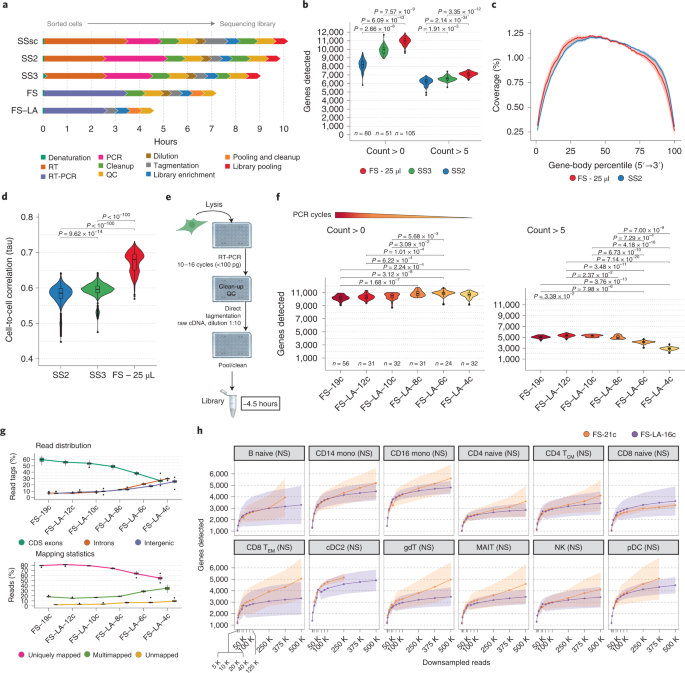
Single-cell RNA-seq (scRNA-seq) has transformed genomics in just a decade. Besides oil-in-emulsion methods for interrogating thousands of cells in parallel, plate-based methods still deserve their own niche because of their superior data quality despite the lower throughput. The most famous among them is perhaps Smart-seq2 which has been adopted by many researchers around the world1. However, fast technological advancements over the years have radically changed experimental needs. New methods like Smart-seq3 now enable single-cell sequencing at a fraction of the original cost while detecting many more genes than Smart-seq22. Despite their usefulness, even the latest plate-based methods still have important drawbacks compared to oil-in-emulsion methods: extensive hands-on time, low throughput and higher costs.
FLASH-seq has been designed to answer many of these issues, enabling the generation of full-length scRNA-seq libraries in half a day, at a similar cost as Smart-seq3. At the time of writing, we tested >300 reaction conditions in FLASH-seq by varying buffers, enzymes and additives, with the vast majority of them negatively affecting the reaction or only providing circumstantial benefits. Our results seem to highlight the limits of the reverse transcriptases of retroviral origin used in virtually all scRNA-seq protocols, which are long known to be very inefficient at converting mRNA into cDNA3,4. However, retroviruses are only one of several classes of retroelements found in nature. Retroviruses evolved to evade host responses by encoding reverse transcriptases with high error rates and low processivity which favours RNA recombination, thus introducing and propagating variations5. Other classes of retroelements, such as prokaryotic non-LTR-retrotransposons and mobile group II introns, evolved the ability to synthesise long cDNA molecules with high fidelity instead. They represent an extraordinary opportunity for single-cell genomics but some drawbacks, such as a low sensitivity and strong binding to the nascent cDNA strand, have precluded their applicability so far6,7. Until these problems are solved, these enzymes are unfortunately not going to replace the retroviral ones for single-cell applications.
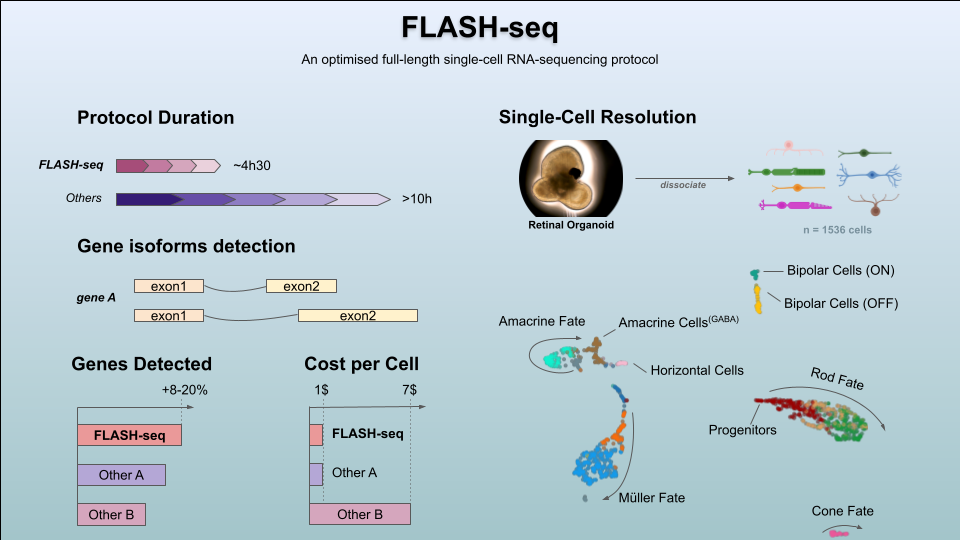
The regular FLASH-seq protocol (‘FLASH-seq’) can be considered an improvement over Smart-seq2. As such, gene counts are the reflection of PCR duplicates. To showcase the flexibility of our protocol, we implemented a random stretch of nucleotides in the TSO sequence (‘FLASH-seq-UMI’), similarly to what was previously described2. These sequences, called Unique Molecular Identifiers (UMIs), are used to barcode every cDNA molecule during RT, by adding a unique tag at the 5’-end. Due to the nature of library preparation which makes use of the Tn5 transposase generating short sequencing amplicons, the final data contains 2 types of reads: terminal reads with the UMI sequence; and internal reads without it. By performing Paired End (PE) sequencing, it is therefore possible to pair each UMI at the 5’-end with internal sequences along the molecule (depending on at which location the Tn5 cut occurred), thus enabling a bioinformatic reconstruction of gene isoforms even with short reads. The balance between internal and UMI reads is a key part of the in silico isoform reconstruction and depends on the amount of forward primer, Tn5 concentration and library input cDNA. However, it is generally difficult to strike a balance between these parameters ahead of sequencing. Given the non-negligible costs of sequencing, such protocols would surely benefit from new quick assays to assess the proportion of UMI reads in a library, for instance by nanopore sequencing or qPCR. While working on FLASH-seq-UMI we demonstrated that the close proximity of the UMI and terminal rGrGrG sequence (i.e. Smart-seq3 TSO) exacerbates the well-known, but often neglected, phenomenon of “strand invasion”. This occurs when the TSO binds to an internal sequence of the RNA or cDNA instead of at the 5’ end, thus creating an artificially truncated cDNA molecule8. The addition of a spacer isolating the two sequences significantly reduced the occurrence of these events in FLASH-seq. The sequence of this spacer should be minimally represented in the transcriptome of interest3 and short enough to limit the UMI inflation4. These observations highlight once again one of the weaknesses of the current scRNA-seq protocols8,9 and the need to visually inspect the data with a genome browser (e.g. Integrated Genome Viewer) for abnormal read distributions when designing new protocols or validating results.
Full-length scRNA-seq is now a mature technology almost on par with emulsion-droplet methods thanks to reduced costs associated with library preparation and sequencing. However, the main limitation remains the number of cells processed in parallel which is constrained by the number of individual wells of the vessel used. Our answer to the throughput problem was the ‘FLASH-seq low-amplification’ protocol. By limiting the cDNA pre-amplification to a few PCR cycles, we were able to perform a direct tagmentation on the cDNA product without any need of intermediate clean-up, cDNA dilution or quality controls. This approach decreased the protocol duration from single cells to sequencing-ready libraries to 4h30, with hands-on-time below 1h. While still compatible with manual pipetting, the protocol can be easily automated to process >1536 cells per day.
We foresee two technological avenues that could drastically increase the throughput of plate-based methods. In the short term, high-density arrays may replace the current 384-well plates. Combined with simplified protocols3,4, the throughput could be increased to several thousands of cells per day. Although similar solutions are already commercially available (I-CELL8 platform from Takara Bio), they still rely on the Poisson distribution for cell deposition, with the results that ⅔ of the wells do not receive a cell. Alternatively, throughput could be increased by performing long-read sequencing, by barcoding individual RNA molecules early on with suitable adaptors and skipping any fragmentation/tagmentation step. This would make the choice of the reaction vessel as well as of automation obsolete. Cells could be captured by using commercially available microwell arrays (BD Rhapsody, Honeycomb Biotechnologies, Singleron Biotechnologies, etc.) or emulsion droplets (10x Genomics, Drop-seq, etc.). Although long-read sequencing has made huge leaps forward in the last few years, costs, data quality and/or throughput are still not competitive with Illumina short-read sequencing. Regardless of the sequencing platform used, an efficient single-cell sequencing method is the result of the optimal combination of enzymes and reagents and we are confident that the introduction of FLASH-seq represents an important contribution to the field.
Finally, we wanted to emphasise two further aspects that are worth being taken into account when designing new protocols: reproducibility and simplicity. New methods often fail to provide sufficient information to the end user, or require complicated setups that are only accessible to a handful of researchers. We believe that scientists should adopt a more commercial mindset with the end user in mind. This does not stop with protocol publication but should be complemented by providing the resources (e.g., assembled microfluidics devices, reagents/protocols sharing, FAQ, etc.) that will make their method a “best-seller”. One step in this direction is the Protocols.io website which is a formidable platform for distributing and maintaining protocols, allowing users and developers to actively engage with each other10-12.
References
- Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
- Hagemann-Jensen, M. et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020).
- Hahaut, V. et al. Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat. Biotechnol. (2022) doi:10.1038/s41587-022-01312-3.
- Hagemann-Jensen, M., Ziegenhain, C. & Sandberg, R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat. Biotechnol. (2022) doi:10.1038/s41587-022-01311-4.
- Mohr, S. et al. Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA N. Y. N 19, 958–970 (2013).
- Belfort, M. & Lambowitz, A. M. Group II Intron RNPs and Reverse Transcriptases: From Retroelements to Research Tools. Cold Spring Harb. Perspect. Biol. 11, a032375 (2019).
- Xu, H., Nottingham, R. M. & Lambowitz, A. M. TGIRT-seq Protocol for the Comprehensive Profiling of Coding and Non-coding RNA Biotypes in Cellular, Extracellular Vesicle, and Plasma RNAs. Bio-Protoc. 11, e4239 (2021).
- Tang, D. T. P. et al. Suppression of artifacts and barcode bias in high-throughput transcriptome analyses utilizing template switching. Nucleic Acids Res. 41, e44 (2013).
- Interpreting Intronic and Antisense Reads in 10x Genomics Single Cell Gene Expression Data.
- Picelli, S. & Hahaut, V. FLASH-seq protocol v3. doi:dx.doi.org/10.17504/protocols.io.kxygxzkrwv8j/v3.
- Picelli, S. & Hahaut, V. FLASH-seq UMI protocol v3. doi:dx.doi.org/10.17504/protocols.io.bp2l619rdvqe/v3.
- Picelli, S. & Hahaut, V. FLASH-seq Low-Amplification protocol v3. doi:https://www.protocols.io/view/flash-seq-umi-protocol-bp2l619rdvqe/v3.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in